Clear Sky Science · de
Vergleichende Bewertung der HTG- und TempO-Seq-Methoden zur gezielten Transkriptom‑Profilierung
Warum das für die Krebsversorgung wichtig ist
Wenn Ärzte und Wissenschaftler Krebs untersuchen, schauen sie häufig auf die „Botenmoleküle“ der Zelle — die RNA — um zu erkennen, welche Gene aktiv oder stillgelegt sind. Solche Muster können zeigen, wie ein Tumor sich verhält und welche Behandlungen am erfolgversprechendsten sind. Die meisten Klinikproben werden jedoch nach Formalinbehandlung in Paraffin eingebettet, was die empfindliche RNA beschädigt. Diese Studie stellt eine praktische Frage mit weitreichenden Folgen für die Krebsforschung: Da ein weit verbreiteter RNA‑Test nicht mehr verfügbar ist, kann eine neuere Methode einspringen und aus diesen routinemäßig konservierten Proben vergleichbar nützliche Ergebnisse liefern?
Zwei Werkzeuge zum Lesen der Genaktivität
Viele Labore nutzten lange das Verfahren HTG EdgeSeq Human Transcriptome Panel (HTP), um die Genaktivität direkt aus kleinen Proben formalinfixierten, paraffineingebetteten (FFPE) Gewebes zu lesen. Dieser Ansatz konnte nahezu alle menschlichen Gene abdecken, ohne dass zuerst RNA extrahiert werden musste, was Zeit sparte und wertvolles Material schützte. Das Unternehmen hinter HTG EdgeSeq meldete jedoch Insolvenz an, sodass Forscher nach Alternativen suchten. Eine neuere Technologie, TempO‑Seq (TOS), von einem anderen Hersteller verspricht ähnliche Fähigkeiten: Sie zielt ebenfalls auf viele Gene gleichzeitig ab, funktioniert mit geschädigter RNA aus FFPE‑Proben und ist auf Sensitivität, Reproduzierbarkeit und relativ niedrige Kosten ausgelegt.
Wie die Methoden gegeneinander getestet wurden
Das Forscherteam verglich diese beiden Technologien direkt in einem sehr praxisnahen Setting. Sie analysierten 21 gelagerte Endometriumkarzinomproben sowie drei standardisierte RNA‑Referenzmaterialien, zuerst mit HTG HTP und dann mit TempO‑Seq. Beide Methoden verwendeten gezielte Panels, die zusammen mehr als 18.000 derselben Gene abdeckten. Die Wissenschaftler unterzogen die Daten strengen Qualitätskontrollen, um sicherzustellen, dass jede Probe genügend Sequenzierungsreads lieferte und die Messungen stabil waren. Außerdem nutzten sie statistische Werkzeuge, um ‚Batch‑Effekte‘ zu entfernen — künstliche Unterschiede, die allein dadurch entstehen können, dass Tests an verschiedenen Tagen, Maschinen oder auf unterschiedlichen Plattformen durchgeführt wurden.
Was übereinstimmt und was nicht
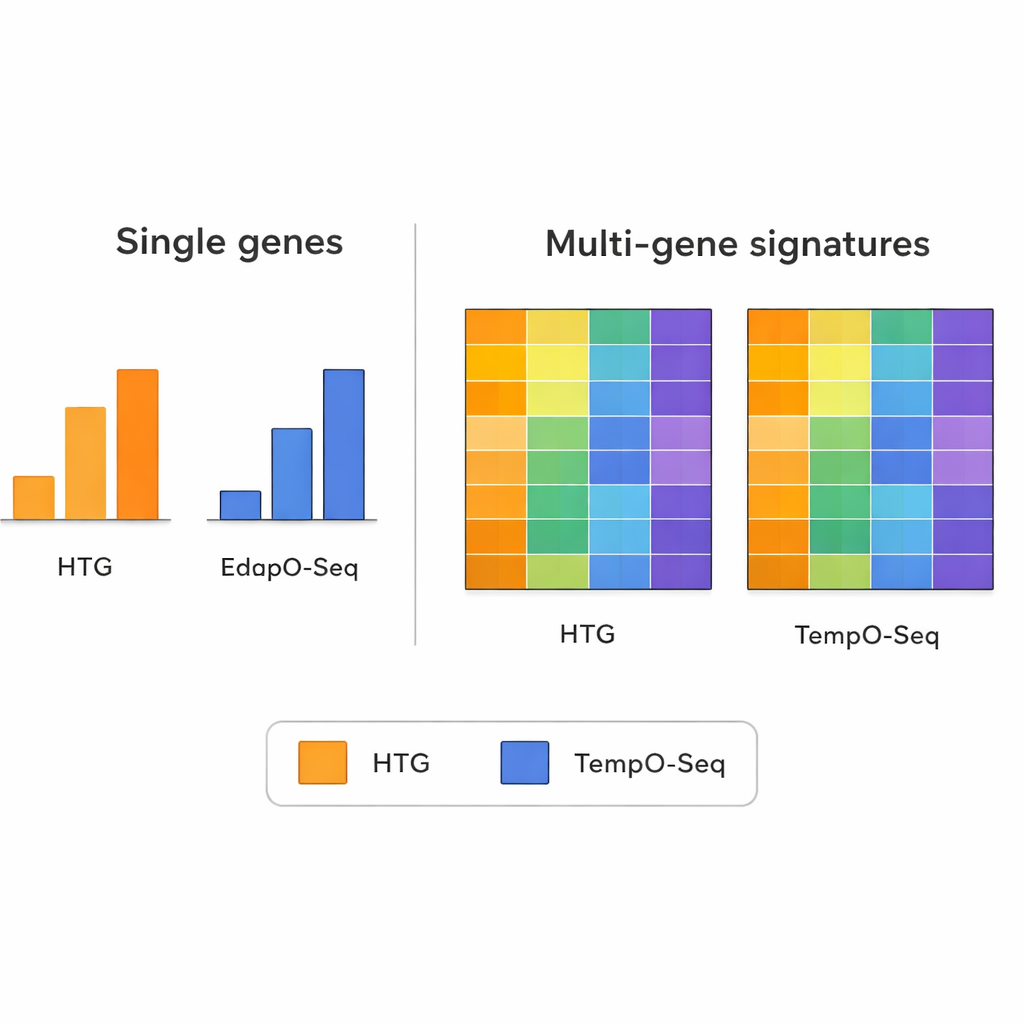
Bei der Betrachtung einzelner Gene zeigten die beiden Methoden nicht immer Übereinstimmung. Unterschiede in der Probe‑Design‑Strategie, der Probenvorbereitung und der Zählweise der Reads können Einzelgenvergleiche verrauschen lassen. Dieses Bild änderte sich jedoch, als die Forscher breitere Muster betrachteten, die Informationen vieler Gene zusammenfassen. Multi‑Gen‑Signaturen — etwa solche, die Tumoren in molekulare Subtypen einteilen, die Anwesenheit von Immunzellen in einer Probe abschätzen oder die Tumorreinheit schätzen — wiesen deutlich stärkere Übereinstimmung zwischen TempO‑Seq und HTG auf. In den meisten Fällen waren die Scores oder Klassifikationen ähnlich, selbst nachdem die Wissenschaftler eine Reduktion der Sequenzierungsreads simulierten, um unterschiedliche Maschinenkapazitäten nachzubilden.
Multi‑Gen‑Muster als verlässliche Signale
Die Studie unterstreicht ein wichtiges Prinzip der modernen Genomik: Während die Messung eines einzelnen Gens durch technische Eigenheiten beeinflusst werden kann, gleicht die Kombination von Signalen aus Dutzenden oder Hunderten Genen dieses Rauschen oft aus. Die Autoren nutzten mehrere bekannte Multi‑Gen‑Werkzeuge als technische Belastungstests. Dazu gehörte ein Brustkrebs‑Panel, das Tumoren intrinsischen Subtypen zuordnet, ein Algorithmus, der misst, wie viel Immun‑ und Bindegewebe in einer Tumorprobe vorhanden ist, und eine Methode zur Abschätzung der Anteile vieler Immunzelltypen. Über diese komplexen Auswertungen hinweg verfolgte TempO‑Seq meist eng die Ergebnisse von HTG, was darauf hindeutet, dass es dieselben biologischen Aussagen erfasst, auch wenn einige feine Details variieren.
Was das für die Zukunft bedeutet
Für Forscher, die auf FFPE‑Archive angewiesen sind, um Krebs zu untersuchen, hätte der Verlust einer bewährten Plattform einen großen Rückschlag bedeuten können. Diese Benchmark‑Studie gibt Zuversicht: TempO‑Seq scheint ein guter Ersatz für HTG HTP zu sein, wenn das Ziel die Nutzung von Multi‑Gen‑Biomarkern und breiten Expressionsmustern ist — die Grundlage vieler moderner diagnostischer und prognostischer Werkzeuge. Die Autoren warnen davor, Einzelgen‑Ergebnisse plattformübergreifend direkt zu vergleichen, da jede Methode Gene auf leicht unterschiedliche Weise erfasst. Stattdessen empfehlen sie, sich bei plattformübergreifender Arbeit auf komplexe Multi‑Gen‑Signaturen zu konzentrieren. Einfach gesagt: Die neue Methode scheint den Job ihres Vorgängers für die meisten praxisnahen onkologischen Forschungsfragen fortführen zu können, besonders wenn es um das Gesamtmuster vieler Gene und nicht den exakten Wert eines einzelnen geht.
Zitation: Fernández-Serra, A., López-Reig, R., Romero, I. et al. Comparative evaluation of HTG and TempO Seq targeted transcriptome profiling methods. Sci Rep 16, 6108 (2026). https://doi.org/10.1038/s41598-026-36810-w
Schlüsselwörter: transkriptomische Profilierung, Endometriumkarzinom, FFPE‑Gewebe, gezielte RNA‑Sequenzierung, Genexpressions‑Biomarker