Clear Sky Science · de
Ein Machine‑Learning‑Ansatz zur Vorhersage osmotischer Koeffizienten und zur Ableitung von Aktivitätskoeffizienten in Alkylammoniumsalzen
Alltägliche Chemikalien mit verborgener Komplexität
Von Weichspülern und Haarspülungen bis zu Desinfektionstüchern und Mundwasser: Eine Gruppe von Verbindungen, die als quartäre Ammoniumsalze bezeichnet werden – oft kurz „Quats“ genannt – steckt unauffällig in vielen Produkten, auf die wir angewiesen sind. Sie helfen, Keime abzutöten, Kleidung zu erweichen und industrielle Reaktionen zu beschleunigen. Trotzdem ist es überraschend schwierig, genau vorherzusagen, wie sich diese Salze in Wasser verhalten, was die effiziente Entwicklung sichererer und umweltfreundlicherer Formulierungen einschränkt. Diese Studie zeigt, wie modernes maschinelles Lernen aus früheren Messungen lernen kann, um dieses Verhalten flexibler und in vielen Fällen genauer vorherzusagen als traditionelle Modelle.
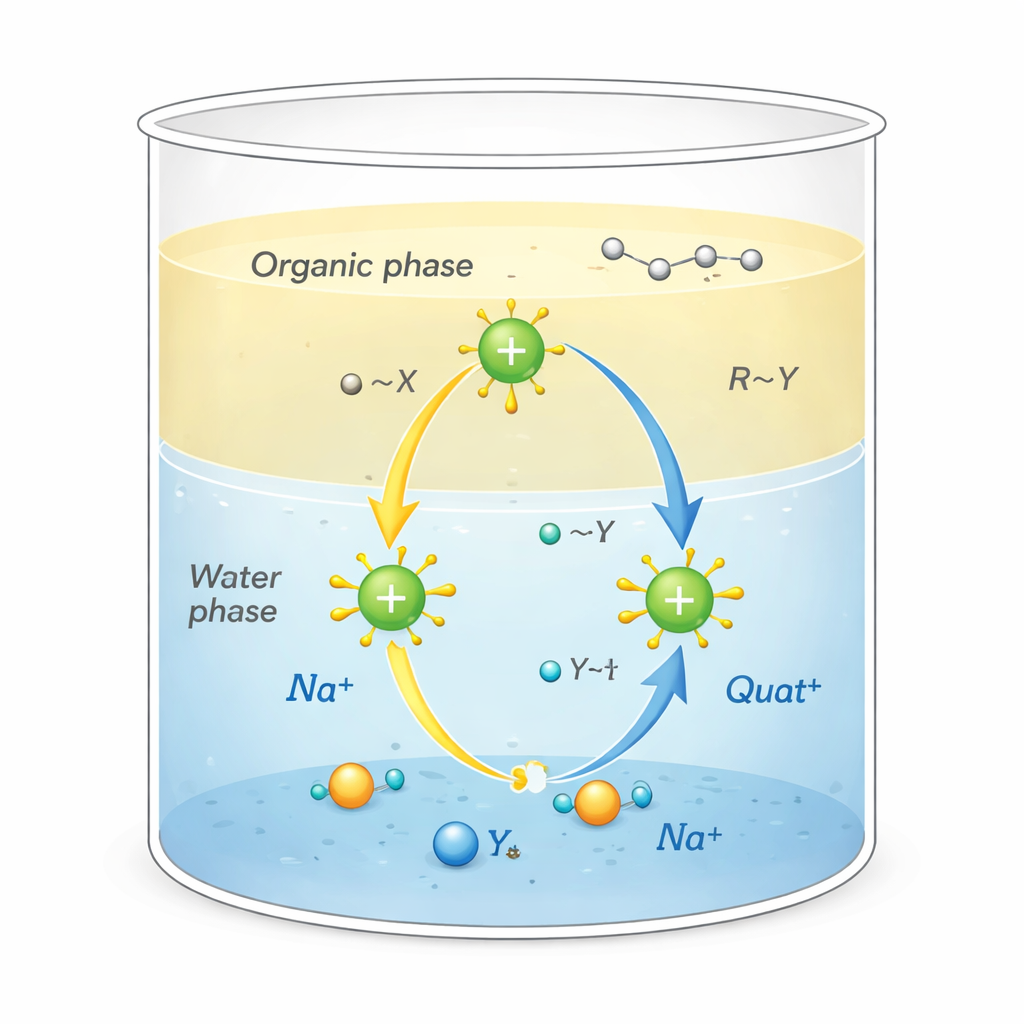
Warum diese Salze wichtig sind
Quats sind positiv geladene Moleküle, umgeben von kohlenstoffreichen „Schwänzen“. Diese ungewöhnliche Form erlaubt ihnen mehrere Aufgaben gleichzeitig: an fettigem Schmutz haften, an Oberflächen wie Stoff oder Haar anhaften und die Membranen von Mikroben stören, wodurch sie wirkungsvolle Desinfektionsmittel und Tenside sind. Sie werden auch als Phasentransferkatalysatoren eingesetzt und fungieren wie Shuttles, die reaktive Ionen aus Wasser in ölähnliche Lösungsmittel transportieren, in die sie sonst nicht gelangen würden. Diese Shuttle‑Wirkung an der Grenze zwischen Wasser und Öl kann chemische Reaktionen, die in der Herstellung von Arzneimitteln, Polymeren und Feinchemikalien verwendet werden, dramatisch beschleunigen.
Warum es schwer ist, ihr Verhalten vorherzusagen
Um neue Quats zu entwerfen oder bestehende anzupassen, müssen Chemiker wissen, wie sie sich in Lösung verhalten – wie stark sie mit Wasser und mit anderen gelösten Ionen wechselwirken. Zwei wichtige Kenngrößen sind der osmotische Koeffizient, der widerspiegelt, wie Salze die Tendenz von Wasser beeinflussen, durch Membranen gezogen zu werden, und der Aktivitätskoeffizient, der erfasst, wie „wirksam“ eine gelöste Spezies im Vergleich zu einer idealen, perfekt gemischten Lösung ist. Traditionell werden diese Werte entweder durch mühsame Experimente oder durch komplexe physikalische Modelle wie Elektrolyte‑NRTL und Extended UNIQUAC gewonnen, die viele angepasste Parameter benötigen und sich nicht leicht auf neue Moleküle verallgemeinern lassen.
Einem Computer das „Lesen“ von Molekülen beibringen
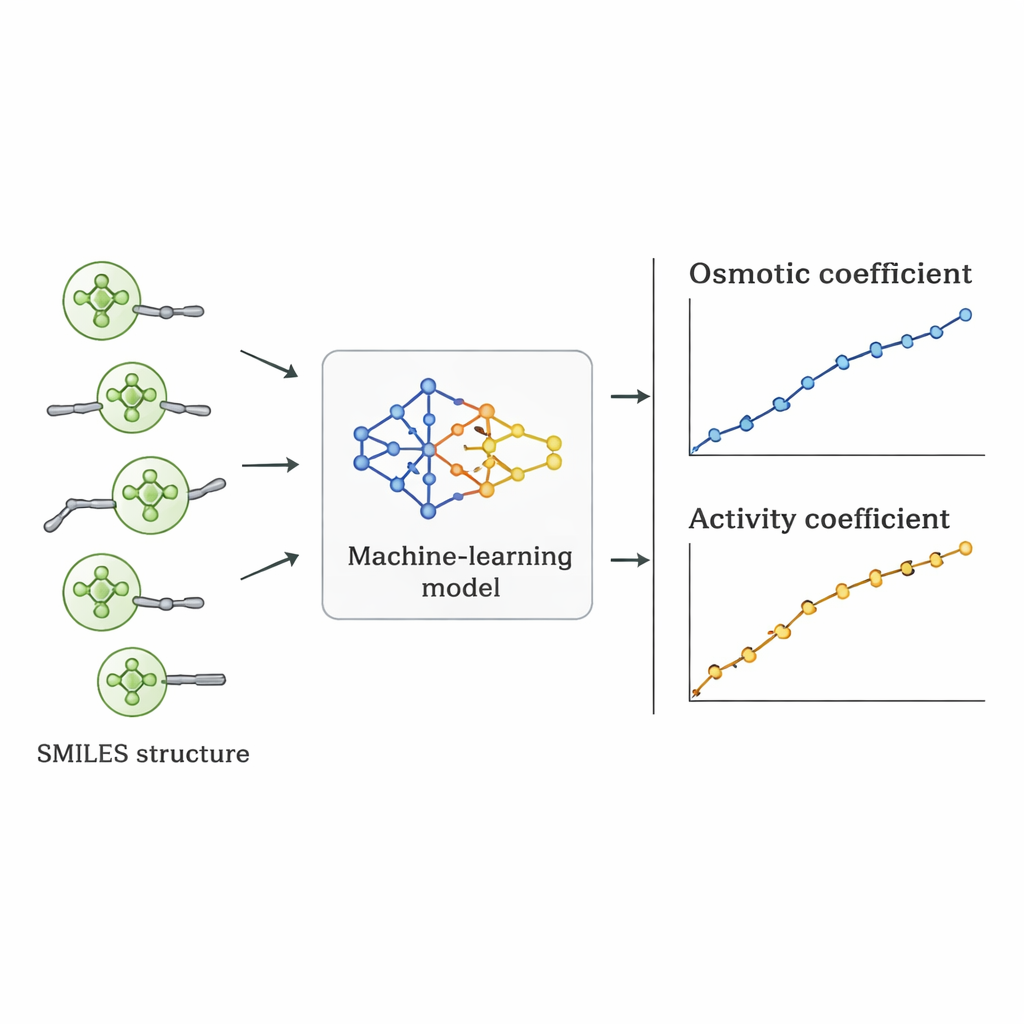
Die Forscher gingen einen anderen Weg: Sie fragten, ob ein Computer die Verbindung zwischen Quat‑Struktur und osmotischem Verhalten direkt aus vorhandenen Daten lernen kann. Sie sammelten 1.654 Messungen osmotischer Koeffizienten für 52 verschiedene Quats aus der Fachliteratur. Jedes Molekül wurde mithilfe der SMILES‑Notation beschrieben – einer Zeichenketten‑Darstellung, die Merkmale wie die Anzahl der Kohlenstoff‑ und Sauerstoffatome, das Vorhandensein von Benzolringen, Verzweigungen und die Art der positiv geladenen Stickstoffgruppe sowie das begleitende negative Ion (z. B. Chlorid, Bromid oder Nitrat) kodiert. Diese Strukturdeskriptoren plus die Salzkonzentration dienten als Eingaben für mehrere überwachte Machine‑Learning‑Algorithmen, die in Python implementiert wurden.
Den zuverlässigsten Prädiktor finden
Sieben verschiedene Algorithmen – darunter lineare Regression, Entscheidungsbäume, Random Forests, Support‑Vector‑Machines, Gradient Boosting, k‑nearest Neighbors und Gaußsche Prozesse – wurden an 70 % der Daten trainiert und an den verbleibenden 30 % getestet. Das Team verwendete außerdem ein strengeres Validierungsschema, bei dem alle Daten für ein Salz zurückgehalten wurden, um zu prüfen, wie gut die Modelle auf wirklich unbekannte Verbindungen extrapolieren. Die lineare Regression schnitt schlecht ab und verfehlte wichtige nichtlineare Trends. Baum‑basierte Methoden passten die Trainingsdaten extrem gut an, lieferten jedoch leicht zackige Vorhersagen und verloren an Genauigkeit bei neuen Salzen. Das Gaußsche Prozessmodell erzielte das beste Gleichgewicht: Es lieferte glatte, physikalisch plausible Kurven für osmotische Koeffizienten und erreichte einen mittleren absoluten prozentualen Fehler von etwa 5 % insgesamt, womit es andere Machine‑Learning‑Ansätze unter den härtesten Prüfungen übertraf.
Vom osmotischen Verhalten zu nützlichen Designzahlen
Sobald das beste Modell ausgewählt war, wurden seine vorhergesagten osmotischen Koeffizienten mithilfe standardmäßiger thermodynamischer Beziehungen in Aktivitätskoeffizienten umgerechnet. Beim Vergleich dieser Aktivitätskoeffizienten mit Werten aus Experimenten und etablierten physikalischen Modellen entsprach der Machine‑Learning‑Ansatz oft oder übertraf diese für einzelne Quats. Obwohl sein mittlerer Fehler über alle Substanzen hinweg etwas höher war als bei einigen spezialisierten Modellen, hatte er einen entscheidenden Vorteil: Da er von Strukturdeskriptoren statt von salzspezifischer Anpassung getrieben wird, lässt er sich auf neue Quats anwenden, die noch nie im Labor gemessen wurden, solange ihre Strukturen denen im Trainingssatz ähneln.
Was das für Produkte und Prozesse bedeutet
Für Nicht‑Spezialisten lautet die Botschaft, dass Computer nun kompakte Textbeschreibungen von Molekülen „lesen“ und aus in der Vergangenheit gelernten Mustern vorhersagen können, wie sich diese Moleküle in Wasser verhalten – mit beeindruckender Genauigkeit. Das eröffnet die Möglichkeit einer schnelleren, kostengünstigeren Vorauswahl neuer Quats für Desinfektionsmittel, Reinigungsmittel, Körperpflegeprodukte und industrielle Katalysatoren, ohne für jede Option exhaustive Experimente durchführen zu müssen. Das aktuelle Modell ist nur ein erster Schritt, und die Autoren weisen darauf hin, dass reichhaltigere molekulare Fingerprints und neuere Algorithmen die Leistung weiter verbessern könnten. Gleichwohl zeigt es, wie datengetriebene Werkzeuge die traditionelle Chemie ergänzen können, indem sie Ingenieuren helfen, effektivere und potenziell sicherere Formulierungen zu entwerfen, indem sie chemische Möglichkeiten erkunden, die im Labor einzeln praktisch nicht testbar wären.
Zitation: Chawuthai, R., Murathathunyaluk, S., Saengsuradech, S. et al. A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts. Sci Rep 16, 5969 (2026). https://doi.org/10.1038/s41598-026-36758-x
Schlüsselwörter: quartäre Ammoniumsalze, Phasentransferkatalyse, osmotische Koeffizienten, Aktivitätskoeffizienten, Maschinelles Lernen in der Chemie