Clear Sky Science · de
ATF4 reguliert mitochondriale Dysfunktion und Mitophagie und trägt so zur Apoptose des Hornhautendothels bei
Warum das Fenster des Auges trübe werden kann
Unsere Hornhäute – die klaren Vorderfenster der Augen – bleiben dank einer dünnen, hart arbeitenden Zellschicht auf ihrer Innenseite transparent. Bei der Fuchs-Endotheldystrophie (FECD) verlieren Millionen von Menschen diese Zellen langsam, was zu Schwellungen, verschwommenem Sehen und häufigen Hornhauttransplantationen führt. Diese Studie stellt eine grundlegende, aber entscheidende Frage: Was veranlasst diese Zellen, den Weg in den Zelltod zu wählen, und könnte das Abschalten eines molekularen „Schalters“ helfen, sie zu retten?
Eine fragile Zellschicht, die das Sehen klar hält
Das Hornhautendothel ist eine einzelne Schicht hexagonaler Zellen, die ständig Flüssigkeit aus der Hornhaut hinauspumpen, um sie klar zu halten. Bei FECD geraten diese Zellen unter Stress und gehen allmählich verloren, während sich Klumpen abnormer Substanz, sogenannte Guttae, auf der darunterliegenden Membran ansammeln. Da es keine zugelassenen Medikamente für FECD gibt und Hornhauttransplantationen die Hauptbehandlung darstellen, versuchen Forschende genau zu verstehen, wie Stress in diesen Zellen sie in Richtung Zelltod treibt. Frühere Arbeiten deuteten getrennt auf Belastungen in zwei wichtigen Zellkompartimenten hin – dem endoplasmatischen Retikulum (der Proteinfaltungsfabrik der Zelle) und den Mitochondrien (den Kraftwerken der Zelle) – doch wie diese beiden Stressantworten miteinander kommunizieren, war unklar.
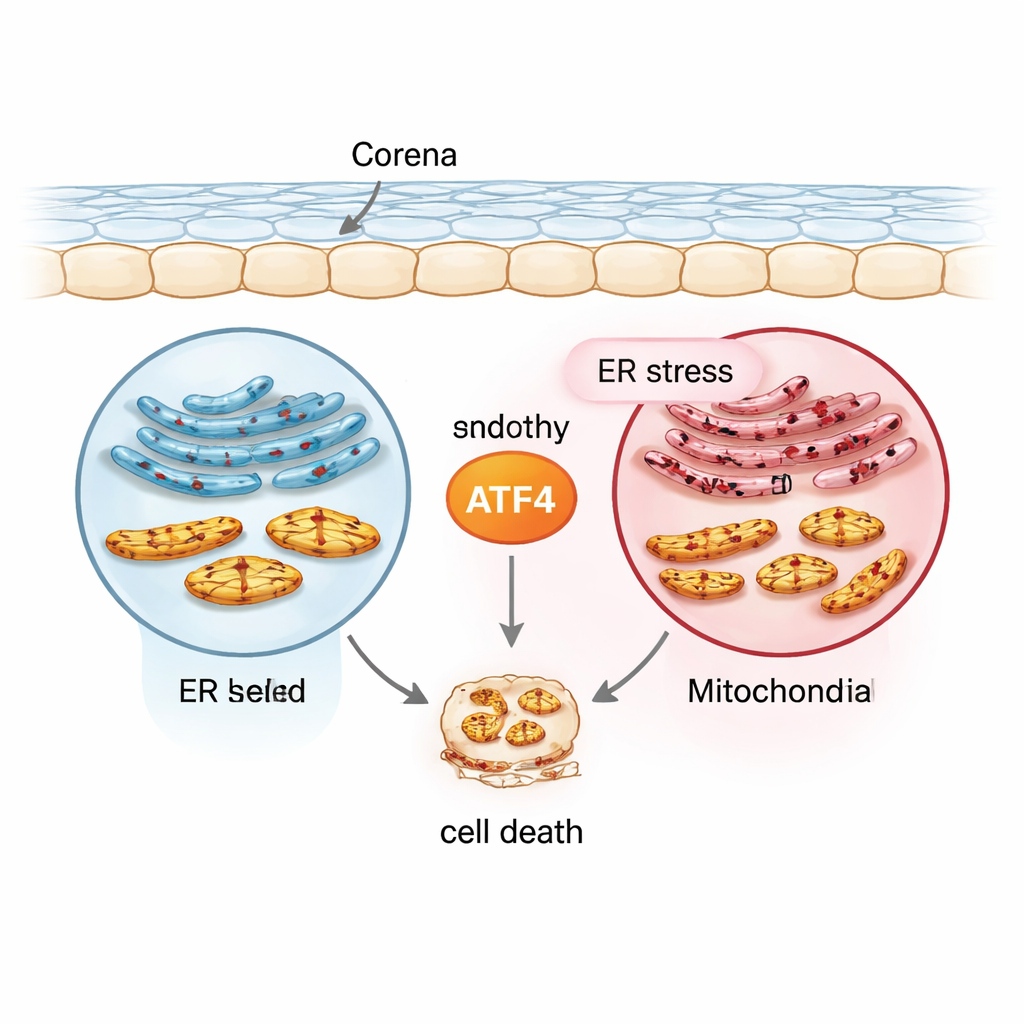
Der Stressbote im Zentrum: ATF4
Das Team konzentrierte sich auf ein Protein namens ATF4, einen Transkriptionsfaktor, der viele Stressantwortgene an- oder abschaltet. Unter Verwendung einer normalen humanen Hornhautendothelzelllinie (21T), einer FECD-ähnlichen Zelllinie mit der krankheitsassoziierten TCF4-Repeat-Expansion (F35T), primärer humaner Hornhautendothelzellen und Mausmodellen, die ultraviolettem A-Licht (UVA) ausgesetzt wurden, schufen sie ein Spektrum von Bedingungen, die chronischen Stress nachahmen. Sie lösten ER-Stress mit dem Wirkstoff Tunicamycin aus und bestimmten dann ATF4 und weitere Marker. Im Vergleich zu normalen Zellen zeigten FECD-ähnliche Zellen bereits zu Beginn höhere Spiegel von ATF4 und verwandten Stressproteinen, und ATF4 stieg unter chronischem Stress sowohl in kultivierten Zellen als auch in menschlichem Hornhautgewebe weiter an. Dieses Muster platzierte ATF4 an der Schnittstelle zwischen frühen Schutzreaktionen und späteren, selbstschädigenden Signalen.
Vom Stromausfall zum programmierten Tod
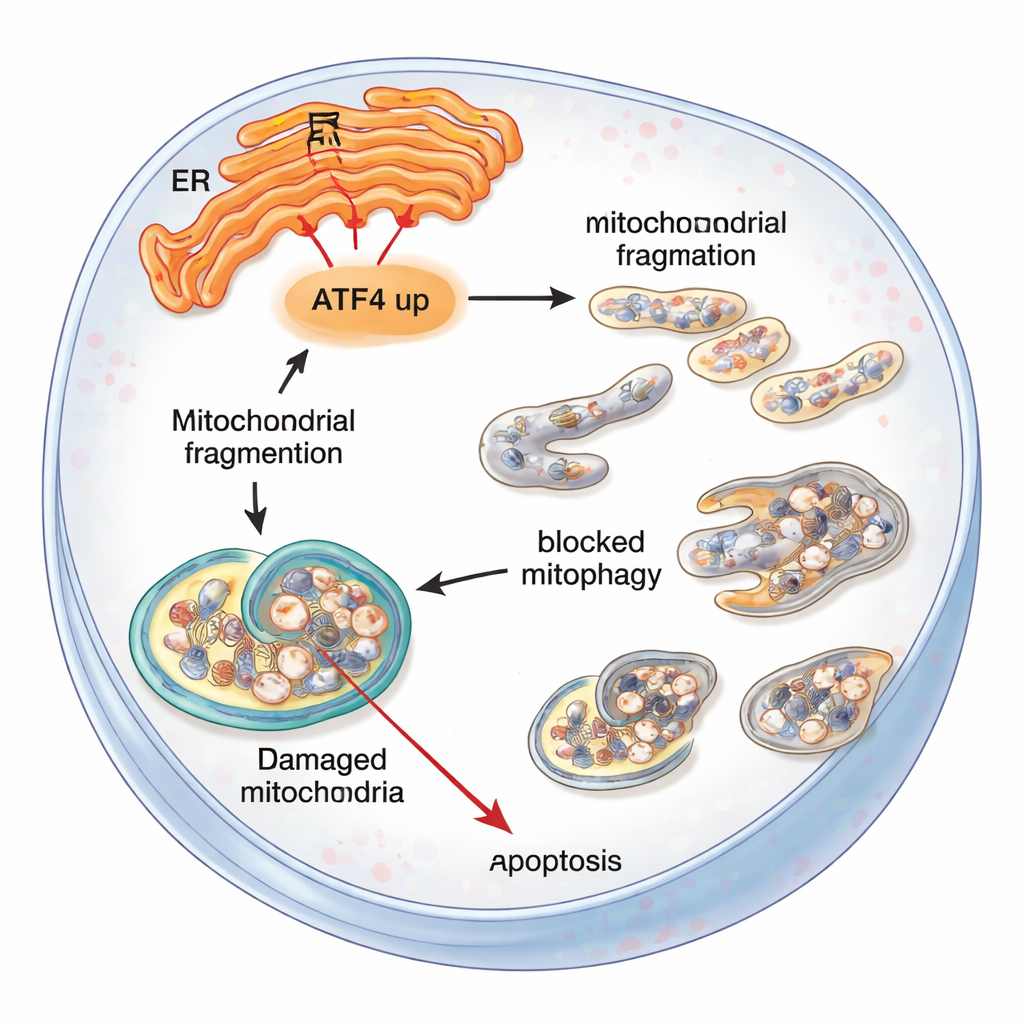
Als Nächstes untersuchten die Forschenden, wie sich dieser Stress auf die Mitochondrien auswirkte. In FECD-ähnlichen Zellen produzierten die Mitochondrien weniger ATP, verloren ihr elektrisches Membranpotenzial und zerfielen von langen, netzwerkartigen Strukturen in viele kleine Fragmente. Diese Veränderungen verschlechterten sich, wenn der ER-Stress länger andauerte. Gleichzeitig wurden klassische Zellsterbeproteine – wie aktivierte Caspasen und das DNA-Reparaturprotein PARP in seiner gespaltenen, pro-apoptotischen Form – häufiger, während schützende Proteine wie Bcl-2 abnahmen. Zusammengenommen deuten diese Veränderungen darauf hin, dass unter Stress stehende Hornhautendothelzellen bei FECD in Richtung einer mitochondriengetriebenen Apoptose gedrängt werden, einer ordentlichen, aber irreversiblen Form des programmierten Zelltods.
Das Aufräumsystem stottert unter chronischem Stress
Normalerweise werden stark beschädigte Mitochondrien durch einen Recyclingmechanismus namens Mitophagie entfernt, bei dem sie markiert und in kleine Vesikel verpackt zur Entsorgung geliefert werden. Das Team fand heraus, dass frühe Mitophagie-Startermoleküle (Parkin und LC3) sowohl in normalen als auch in FECD-ähnlichen Zellen aktiviert wurden, besonders nach Stress. Allerdings waren wichtige unterstützende Proteine reduziert, und die Elektronenmikroskopie zeigte eine Anhäufung teilweise verdauter Mitochondrien, die in Vesikeln gefangen waren. Das legt nahe, dass der Aufräumprozess zwar begonnen wurde, aber nicht abgeschlossen werden konnte, sodass die Zellen mit defekten Kraftwerken überfrachtet blieben, was den Stress und den Zelltod weiter anheizt statt einer Erholung zu dienen.
ATF4 ausschalten, um Zellen zu retten
Um zu prüfen, ob ATF4 diesen Teufelskreis antreibt, setzten die Forschenden small interfering RNA ein, um ATF4 in kultivierten Hornhautendothelzellen teilweise zu stilllegen. Unter demselben chronischen Stress zeigten Zellen mit reduziertem ATF4 geringere Spiegel an den Tod fördernden Proteinen, ein gesünderes mitochondriales Membranpotenzial, weniger Fragmentierung und bessere Überlebensraten in Vitalitätstests. Wichtig war, dass die Anzahl feststeckender Mitophagie-Strukturen abnahm, was darauf hindeutet, dass eine Senkung von ATF4 dazu beitrug, das Gleichgewicht zwischen Schäden und Reinigung wiederherzustellen. In Mäusen, die so verändert wurden, dass sie nur noch eine funktionierende Kopie des ATF4-Gens besitzen, führte UVA-Exposition zu weniger Aktivierung eines pro-apoptotischen Partnerproteins, CHOP, und bewahrte im Vergleich zu völlig ATF4-ausgestatteten Mäusen eine größere Zahl normal geformter Endothelzellen.
Was das für Menschen mit FECD bedeutet
Für Nichtfachleute lautet die Botschaft: Ein Stressbote, ATF4, kann Hornhautendothelzellen vom Bewältigen in den Zusammenbruch kippen. Wenn ER-Stress langanhaltend ist, stört ATF4 die Mitochondrien, blockiert die Aufräummaschinerie der Zelle und fördert letztlich die Selbstzerstörung dieser lebenswichtigen Zellen. Das Herunterregeln von ATF4 – entweder genetisch in Mäusen oder mit gezielten molekularen Werkzeugen in Zellen – schützt Mitochondrien, verbessert die Abfallentsorgung und erhält mehr Zellen am Leben. Obwohl diese Arbeit noch auf Labor- und Tiermodellen beruht, macht sie ATF4 und verwandte Stresswege zu vielversprechenden Wirkstoffzielen, die eines Tages das Fortschreiten der Fuchs-Dystrophie verlangsamen oder verhindern und so den Bedarf an Hornhauttransplantationen reduzieren könnten.
Zitation: Qureshi, S., Kim, S.Y., Lee, S. et al. ATF4 regulates mitochondrial dysfunction and mitophagy, contributing to corneal endothelial apoptosis. Sci Rep 16, 5960 (2026). https://doi.org/10.1038/s41598-026-36453-x
Schlüsselwörter: Fuchs-Endotheldystrophie der Hornhaut, Hornhautendothel, mitochondrialer Stress, Mitophagie, ATF4