Clear Sky Science · de
Hybrides Deep-Learning-Framework für die präzise Klassifikation hochdimensionaler Genomdaten
Den Datenstrom des Genoms verstehen
Moderne DNA-Technologien können in einem einzigen Experiment Zehntausende Gene messen und versprechen so frühere Krankheitsdetektion und präzisere Therapien. Doch diese Datenfülle ist so groß, verrauscht und komplex, dass selbst leistungsfähige Computermodelle oft Schwierigkeiten haben, klare und vertrauenswürdige Muster zu finden. Dieses Paper stellt ein neues KI-System vor, das speziell dafür entwickelt wurde, mit solchen überwältigenden Genomdaten umzugehen, mit dem Ziel, Vorhersagen genauer zu machen und gleichzeitig zu erklären, wie diese Vorhersagen zustande kamen.
Warum Genomdaten so schwer zu nutzen sind
Genomstudien liefern routinemäßig weit mehr Messwerte als Patienten oder Proben vorhanden sind. Viele dieser Messwerte sind irrelevant, redundant oder durch technische Störungen verzerrt. Traditionelle Machine-Learning-Methoden benötigen entweder Experten, die manuell auswählen, welche Gene relevant sein könnten, oder sie versuchen, alles zu verwenden und riskieren Overfitting — also gute Leistung auf Trainingsdaten, aber Versagen bei neuen Fällen. Deep Learning, das Felder wie die Bilderkennung revolutioniert hat, kann automatisch Muster aus Rohdaten lernen. In der Genomik verhält es sich jedoch häufig wie eine Blackbox: Es liefert möglicherweise genaue Antworten, gibt aber wenig Einblick in die zugrundeliegenden Gründe, was die Akzeptanz in der Medizin einschränkt, wo Transparenz entscheidend ist.
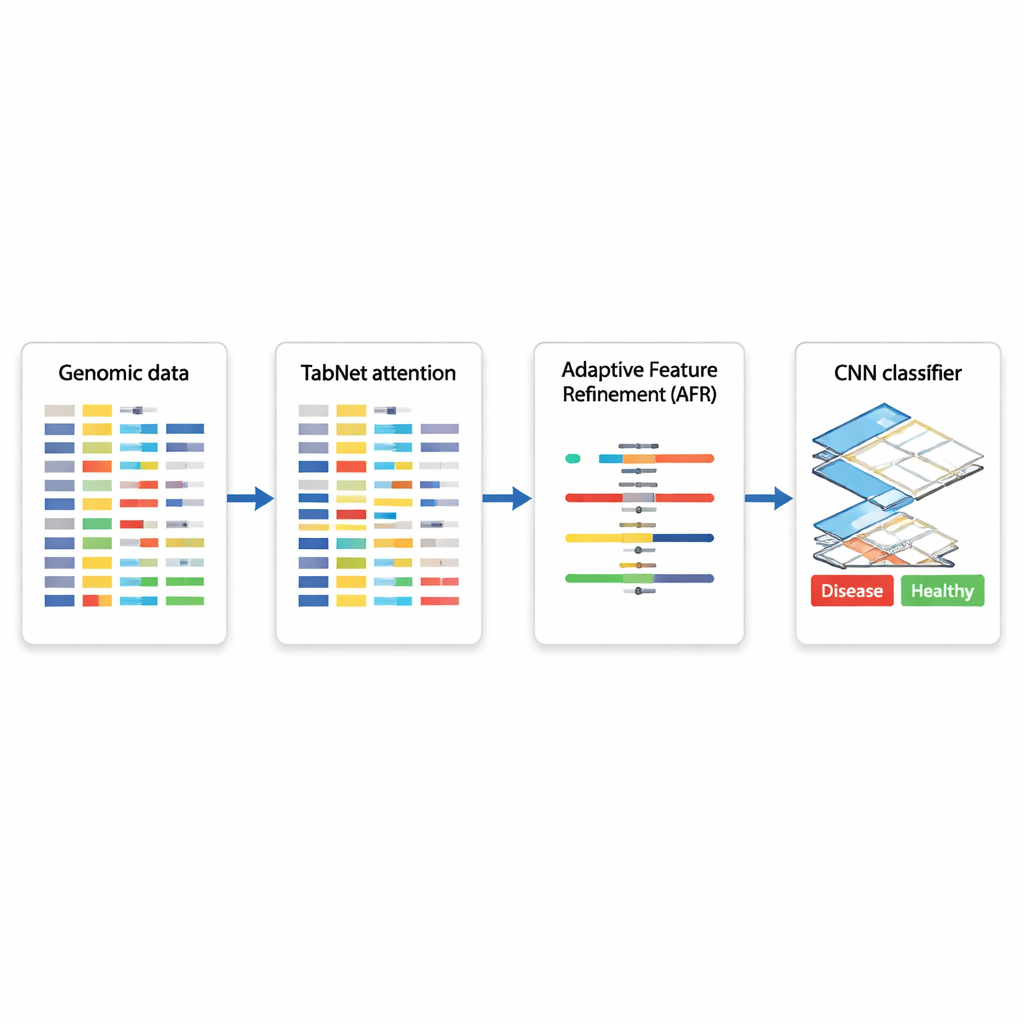
Ein hybrider KI-Entwurf für auf Genen basierende Entscheidungen
Die Autorinnen und Autoren schlagen eine hybride Deep-Learning-Architektur vor, die drei spezialisierte Module hintereinanderschaltet. Zuerst fungiert eine Komponente namens TabNet wie ein Spotlicht, das alle verfügbaren genomischen Messwerte durchscannt und lernt, welche Merkmale für eine gegebene Aufgabe am informativsten sind — zum Beispiel das Unterscheiden von krebsartigem und nicht-krebsartigem Gewebe. Anstatt jedes Gen gleich zu behandeln, richtet TabNet die Aufmerksamkeit auf eine spärliche Teilmenge, die am relevantesten erscheint. Anschließend nimmt eine Adaptive Feature Refinement (AFR)-Schicht diese ausgewählten Signale und gewichtet sie neu, stärkt konsistente, sinnvolle Muster und dämpft weiteres Rauschen. Schließlich untersucht ein Convolutional Neural Network (CNN), wie in der Bildanalyse häufig verwendet, wie die verfeinerten Merkmale lokal interagieren und erfasst so subtile Beziehungen zwischen Gen-Gruppen, die auf einen bestimmten Subtyp einer Krankheit oder einen biologischen Zustand hinweisen könnten.
Das Modell auf die Probe stellen
Das Framework wurde an drei großen öffentlichen Datensätzen evaluiert: einem Brustkrebsdatensatz aus The Cancer Genome Atlas, einem Single-Cell-Melanom-Datensatz aus dem Gene Expression Omnibus und einem Epigenomik-Datensatz aus dem ENCODE-Projekt. Zusammen umfassen diese Sammlungen Tausende von Proben und Zehntausende von Merkmalen pro Probe, die Genaktivität und chemische Markierungen auf der DNA abdecken. Über alle Datensätze hinweg übertraf das hybride Modell mehrere moderne Ansätze und verbesserte die Genauigkeit sowie wichtige Klassifikationsmetriken wie die Fläche unter der Receiver-Operating-Characteristic-Kurve (AUC) und den F1-Score um etwa 5–8 Prozentpunkte. Wichtig ist, dass diese Verbesserungen nicht zulasten der Transparenz gingen: Das Modell liefert Aufmerksamkeitskarten von TabNet und Aktivierungskarten vom CNN, die hervorheben, welche Gene und Regionen bei jeder Vorhersage am einflussreichsten waren.
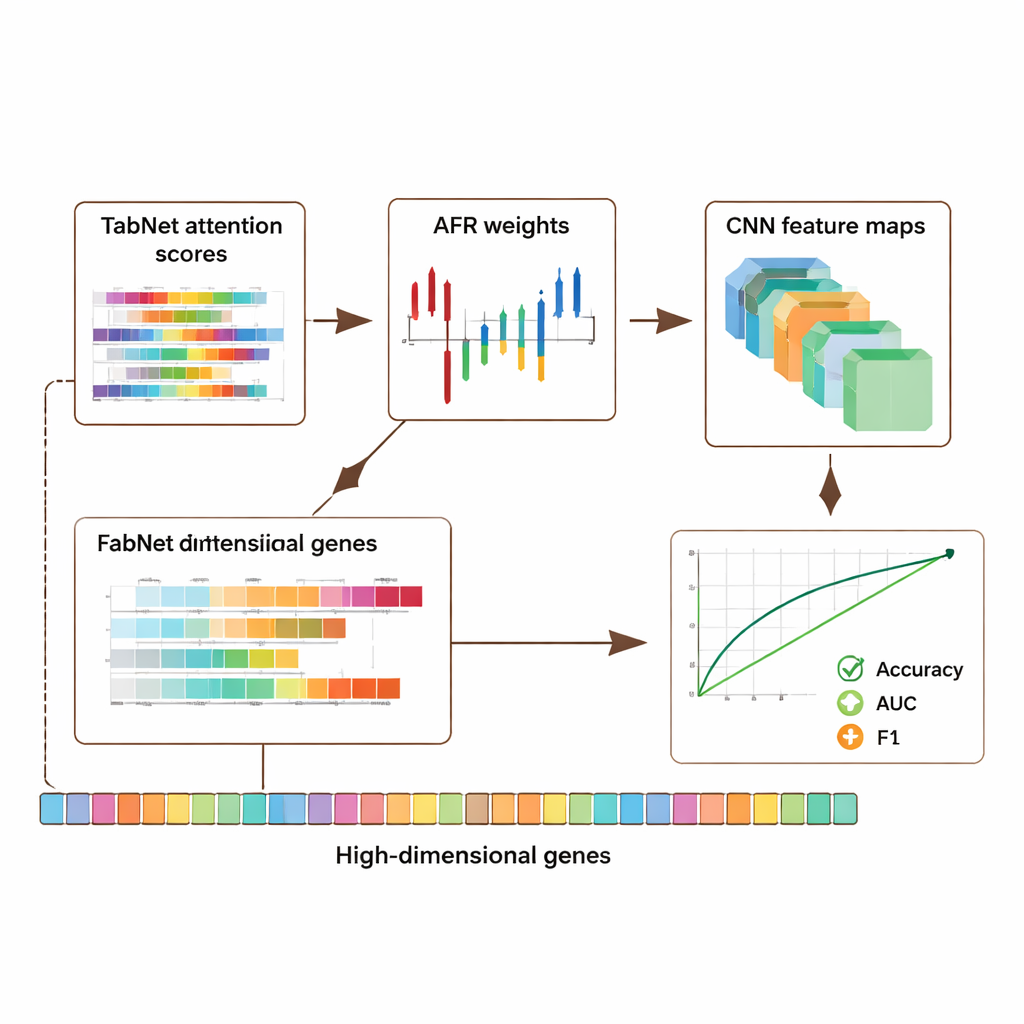
Balance zwischen Genauigkeit, Privatsphäre und Vertrauen
Da Genomdaten hochgradig persönlich sind, untersuchten die Autorinnen und Autoren außerdem, wie man Privatsphäre schützen kann, ohne nützliche Signale zu verlieren. Sie führten einen adaptiven Datenschutzmechanismus ein, der besonders sensiblen Merkmalen mehr Rauschen und anderen weniger Rauschen hinzufügt, kombiniert mit Maskierung ausgewählter Eingaben. Tests zeigten, dass das Modell selbst bei moderatem Rauschen eine starke Genauigkeit und Diskriminationsfähigkeit beibehielt und seine Leistung graduell abnahm, wenn der Schutz erhöht wurde. Gleichzeitig deuteten die interpretierbaren Aufmerksamkeits- und Aktivierungsmuster häufig auf Gene hin, die bereits für Rollen in Krebs und Immunregulation bekannt sind, was darauf hindeutet, dass das System nicht bloß Daten auswendig lernt, sondern biologisch sinnvolle Signale erfasst. Eine Ablationsstudie — das systematische Entfernen von Architekturteilen — bestätigte, dass jedes Modul, insbesondere die AFR-Schicht, einen messbaren Beitrag zur Leistung leistete.
Was das für die Medizin der Zukunft bedeutet
Einfach gesagt bietet diese Arbeit einen intelligenteren Weg, riesige genomische Tabellen nach krankheitsassoziierten Mustern zu durchsuchen und gleichzeitig aufzuzeigen, welche Einträge in der Tabelle am wichtigsten waren. Durch die Kombination gezielter Merkmalsauswahl, sorgfältiger Verfeinerung und Mustererkennung verbessert das hybride Modell die Vorhersagegenauigkeit, bleibt rechnerisch handhabbar und liefert visuelle Hinweise, die Klinikerinnen, Kliniker und Biologinnen und Biologen interpretieren können. Obwohl umfassendere Tests an breiteren und vielfältigeren Patientengruppen noch nötig sind, könnten solche Frameworks helfen, neue Biomarker zu identifizieren, Krankheitsuntertypen zu verfeinern und klinische Entscheidungswerkzeuge in der Präzisionsmedizin zu unterstützen — und so die KI-gestützte DNA-Analyse einen Schritt näher an den realen Einsatz bringen.
Zitation: Swain, M.K., Kamila, N.K., Jena, L. et al. Hybrid deep learning framework for accurate classification of high dimensional genomic data. Sci Rep 16, 5919 (2026). https://doi.org/10.1038/s41598-026-36128-7
Schlüsselwörter: genomisches Deep Learning, Entdeckung von Krebs-Biomarkern, interpretierbare KI, Präzisionsmedizin, datenschutzwahrende Genomik