Clear Sky Science · de
Eine quantitative Studie zytotoxischer Verbindungen mithilfe graphbasierter Deskriptoren und maschinellen Lernens
Warum diese Forschung für künftige Krebsmedikamente wichtig ist
Krebsmedikamente, die Tumorzellen abtöten und als zytotoxische Wirkstoffe bekannt sind, balancieren häufig zwischen Lebensrettung und schweren Nebenwirkungen. Um sicherere und wirksamere Therapien zu entwickeln, benötigen Wissenschaftler schnelle und verlässliche Methoden, um vorherzusagen, wie sich diese Arzneistoffe im Körper verhalten—wie gut sie aufgenommen werden, wie leicht sie Zellmembranen passieren und wo sie sich anreichern. Diese Studie zeigt, wie mathematische Beschreibungen von Wirkstoffmolekülen in Kombination mit modernem maschinellen Lernen eine Schlüssigeigenschaft, die dieses Verhalten steuert, genau abschätzen können und so die Suche nach besseren Krebstherapien beschleunigen könnten.
Eine zentrale Oberfläche, die steuert, wohin Medikamente gelangen
Ein zentraler Begriff der Arbeit ist die topologische polare Oberfläche, kurz Top_PSA. Einfach ausgedrückt ist dies eine Zahl, die widerspiegelt, wie groß der Anteil der „polaren“ Bereiche an der Moleküloberfläche ist—also jene Bereiche, die Wasser mögen und Wasserstoffbrücken bilden können. Moleküle mit sehr hoher polarer Oberfläche haben oft Schwierigkeiten, fettige Zellmembranen zu durchdringen, und können bei oraler Einnahme schlecht aufgenommen werden. Moleküle mit sehr geringer polarer Oberfläche passieren Barrieren dagegen oft zu leicht und können unerwünschte Wirkungen in empfindlichen Geweben wie dem Gehirn haben. Top_PSA hat sich als praktischer Indikator für diese Transporteigenschaften etabliert, weil er schnell aus einer 2D-Zeichnung eines Moleküls berechnet werden kann, ohne langsame 3D-Simulationen zu benötigen.
Molekülzeichnungen in Zahlen verwandeln
Die Forscher stellten einen kuratierten Satz von 156 verschiedenen zytotoxischen Verbindungen zusammen, entnommen aus realen Krebsmedikamenten und experimentellen Substanzen. Sie wandelten jedes Molekül dann in 58 sogenannte Deskriptoren um—Zahlen, die Merkmale erfassen wie die Anzahl der Atome, die Anzahl der Ringe, die Flexibilität der Bindungen, wie viele Atome Wasserstoffbrücken bilden können und wie polar oder elektronegativ einzelne Teile sind. Viele dieser Deskriptoren stammen aus der Graphentheorie, in der ein Molekül als Netzwerk aus verbundenen Knoten und Kanten behandelt wird. Dieses umfassende numerische Porträt jedes Moleküls diente als Eingabe für Computermodelle, die versuchten, die Top_PSA-Werte vorherzusagen, wie sie von weit verbreiteten Chemie-Toolkits berechnet werden.
Mehrere Wege zu genauer Vorhersage testen
Um den besten Weg zu finden, diese Deskriptoren mit Top_PSA zu verknüpfen, verglich das Team mehrere Modellierungsstrategien. Sie testeten einfache lineare Regression sowie zwei „regularisierte“ Versionen, genannt Ridge- und LASSO-Regression, die besser mit verrauschten und überlappenden Informationen umgehen sollen. Außerdem untersuchten sie unterschiedliche Datenvorbereitungs-Schemata: Modelle direkt auf den rohen Deskriptoren anzupassen, diese mit Hauptkomponenten-Analyse (PCA) zu komprimieren, sie mit robustem Scaling zu normieren, Ausreißer zu korrigieren und stark korrelierte Merkmale mithilfe des Variance Inflation Factor zu entfernen. Jede Vorgehensweise wurde sorgfältig mittels k‑facher Kreuzvalidierung bewertet, einer Methode, die die Daten wiederholt in Trainings- und Testmengen aufteilt, um Overfitting zu vermeiden.
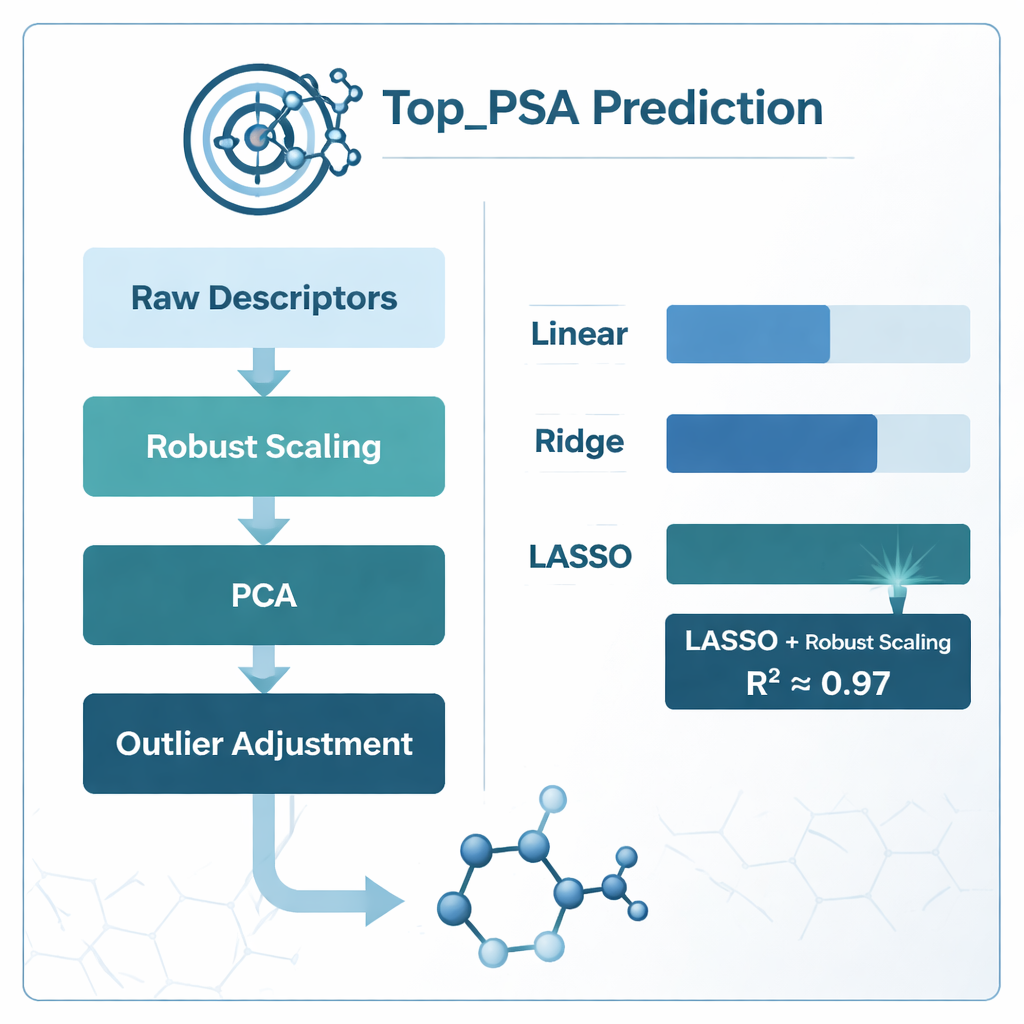
Was am besten funktionierte und was die Modelle lernten
Der klare Gewinner war die Kombination aus robustem Scaling und LASSO-Regression, die einen Bestimmtheitskoeffizienten (R²) von etwa 0,97 erreichte—was bedeutet, dass sie ungefähr 97 % der Variation in Top_PSA über die 156 Verbindungen erklären konnte. Auf PCA basierende Modelle kamen in der Rohgenauigkeit nahe heran, waren chemisch jedoch schwerer zu interpretieren, weil die ursprünglichen Deskriptoren zu abstrakten Komponenten verschmolzen werden. Ein einfaches Entfernen korrelierter Deskriptoren mithilfe des Variance Inflation Factor schadete der Leistung sogar, was darauf hindeutet, dass einige überlappende Messgrößen trotzdem nützliche chemische Informationen tragen. Durch die Analyse der von LASSO aufrechterhaltenen Nicht-Null-Gewichte fanden die Autoren heraus, dass die wichtigsten Faktoren das Vorhandensein von Heteroatomen wie Stickstoff und Sauerstoff, die Fähigkeit, Wasserstoffbrücken zu geben oder zu akzeptieren, und Indizes sind, die verfolgen, wie elektronegative Atome im molekularen Graphen angeordnet sind—alles Merkmale, die dem chemischen Verständnis der polaren Oberfläche entsprechen.
Wie dies die Wirkstoffentwicklung leiten kann
Für Leser außerhalb des Fachgebiets lautet die Kernaussage, dass sorgfältig aufbereitete mathematische Fingerabdrücke von Molekülen in Kombination mit gut gewählten Methoden des maschinellen Lernens schnelle und verlässliche Abschätzungen liefern können, wie „haftend“ oder „gleitend“ Krebsmedikamente auf ihrem Weg durch den Körper sind. Die Studie bietet praktische Hinweise für andere Forscher, wie Deskriptor-Daten vorzubereiten sind, welche Modellierungsansätze zu bevorzugen sind und welche Abkürzungen zu vermeiden sind. Langfristig können solche robusten, interpretierbaren Modelle von Top_PSA Chemikern helfen, riesige virtuelle Bibliotheken potenzieller Wirkstoffe vorzusortieren und die Arbeit auf Verbindungen mit dem richtigen Gleichgewicht zwischen Membrandurchquerung und Sicherheit zu konzentrieren—ein wichtiger Schritt hin zu wirksameren und weniger toxischen Krebstherapien.
Zitation: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Schlüsselwörter: zytotoxische Medikamente, polare Oberfläche, molekulare Deskriptoren, maschinelles Lernen, Wirkstoffpermeabilität