Clear Sky Science · de
Mitochondriale Fehlfunktion und Ca2+-Dysregulation in aus humanen iPS-Zellen abgeleiteten Neuronen mit Presenilin‑1-Mutation treten unter Stress über einen MCU‑1-unabhängigen Mechanismus auf
Warum das für die Alzheimer‑Krankheit wichtig ist
Die Alzheimer‑Krankheit wird oft über klebrige Proteinplaques im Gehirn beschrieben, doch lange bevor das Gedächtnis nachlässt, können die winzigen „Kraftwerke“ in Nervenzellen – die Mitochondrien – und die Handhabung von Calciumionen bereits gestört sein. Diese Studie verwendet menschliche Neuronen, die aus Hautzellen einer Person mit einer bekannten familiären Alzheimer‑Mutation gezüchtet wurden, um eine einfache, aber zentrale Frage zu stellen: Wie früh und auf welche Weise versagen Energieproduktion und Calcium‑Gleichgewicht?
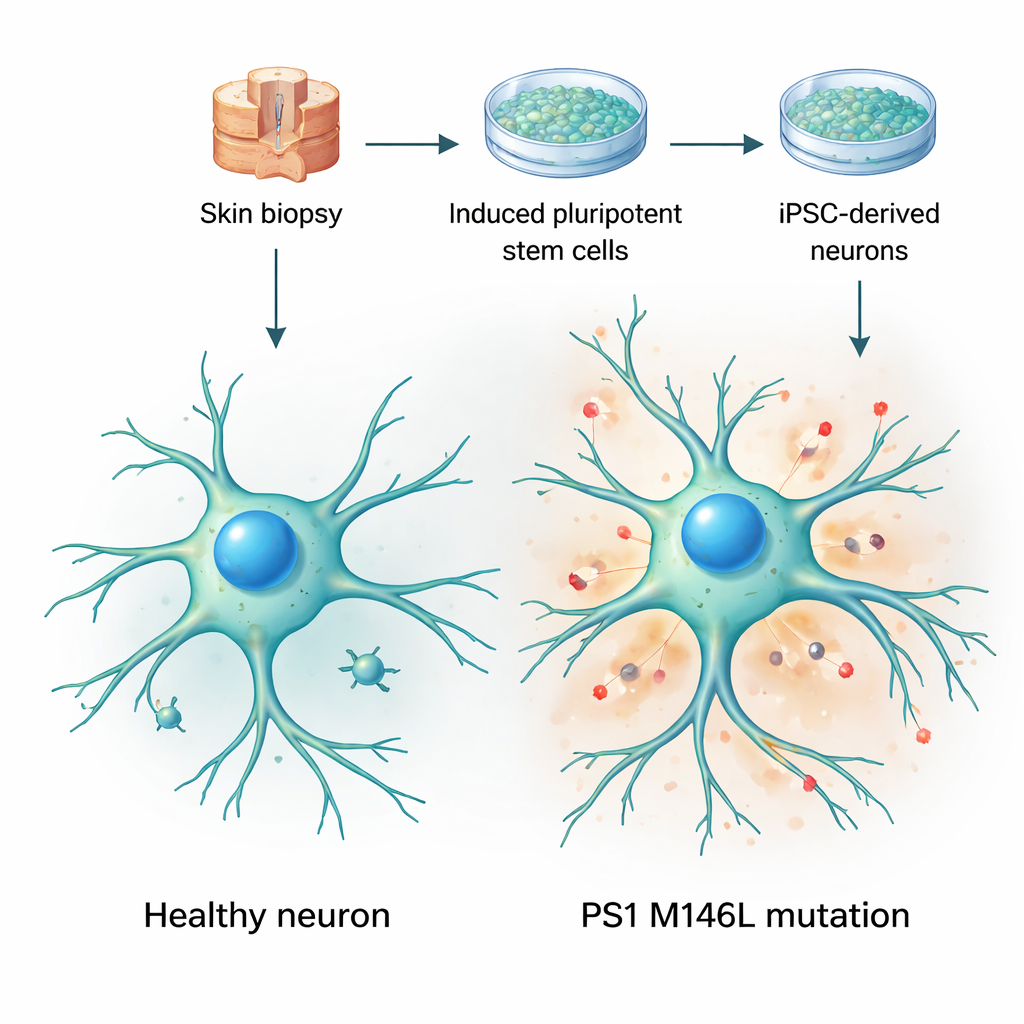
Aus Hautzellen lebende Gehirnmodelle erzeugen
Die Forschenden begannen mit Hautbiopsien von zwei Frauen: einer gesunden Freiwilligen und einer symptomfreien Trägerin der Presenilin‑1‑Mutation M146L, die in einer argentinischen Familie mit früh einsetzender Alzheimer‑Krankheit auftritt. Sie reprogrammierte die Hautzellen zu induzierten pluripotenten Stammzellen – Zellen, die sich fast in jedes Gewebe verwandeln können – und leiteten diese dann zu Nervenzellen. Über mehrere Wochen in Kultur nahmen diese Zellen typische neuronale Formen an, bildeten lange, verzweigte Fortsätze und exprimierten gängige neuronale Marker. Wichtig ist, dass sich sowohl die Kontroll‑ als auch die Mutanten‑Zellen in ähnlichem Tempo entwickelten und allgemein gesund wirkten, was dem Team erlaubte, sich auf subtile funktionelle Veränderungen statt auf offensichtlichen Zellverlust oder Schaden zu konzentrieren.
Elektrische Signale und Calcium unter Belastung
Nervenzellen sind auf eine enge Kontrolle von Calcium angewiesen, einem geladenen Atom, das als schneller Ein/Aus‑Schalter für viele zelluläre Prozesse fungiert. Mit fluoreszierenden Farbstoffen verfolgte das Team, wie sich die Calciumspiegel in den Zellen änderten, wenn diese elektrisch mit Kalium stimuliert oder mit Signalstoffen aktiviert wurden. Unter einfacher depolarisierender Stimulation zeigten Neuronen mit der M146L‑Mutation schwächere Calciumanstiege als Kontrollneuronen, was auf Probleme bei der Aufrechterhaltung der elektrischen und ionischen Gradienten hindeutet, die normalerweise den Calciumeinstrom treiben. Als die Forschenden jedoch eine stressigere Situation auslösten – indem sie Calcium aus internen Speichern des endoplasmatischen Retikulums entweichen ließen – wurde der Unterschied deutlicher. Als Reaktion auf diesen Stress nahmen die Mitochondrien in den mutierten Neuronen merklich weniger Calcium auf als in den Kontrollzellen, was auf eine reduzierte Fähigkeit hinweist, gefährliche Calciumspitzen abzufangen.
Energieverbrauch vom Calciumhaushalt entkoppelt
Um zu verstehen, wie diese veränderte Calciumhandhabung den Stoffwechsel beeinflusst, maßen die Untersuchenden den Sauerstoffverbrauch der Neuronen – ein direkter Proxy für mitochondriale Aktivität. Überraschenderweise atmeten Neuronen mit der M146L‑Mutation stärker: Ihre basalen und maximalen Sauerstoffverbrauchsraten sowie der Teil des Sauerstoffs, der mit ATP‑Produktion verbunden ist, lagen alle höher als in Kontrollzellen. Dennoch schien die Effizienz, mit der Sauerstoffverbrauch in ATP‑Produktion gekoppelt ist, ähnlich zu sein, und es gab keinen Anstieg der Mitochondrienzahl oder der zentralen ATP‑bildenden Enzyme. Stattdessen waren die Mitochondrien in den Mutanten länger und röhrenförmiger und wiesen höhere Mengen eines Fusionsproteins namens Mitofusin‑1 auf – ein Muster, das häufig in Zellen unter chronischem, niedriggradigem Stress beobachtet wird. Diese hyperaktiven, verlängerten Mitochondrien erzeugten außerdem mehr reaktive Sauerstoffspezies, instabile Moleküle, die Proteine und DNA schädigen können, wenn sie nicht kontrolliert werden.
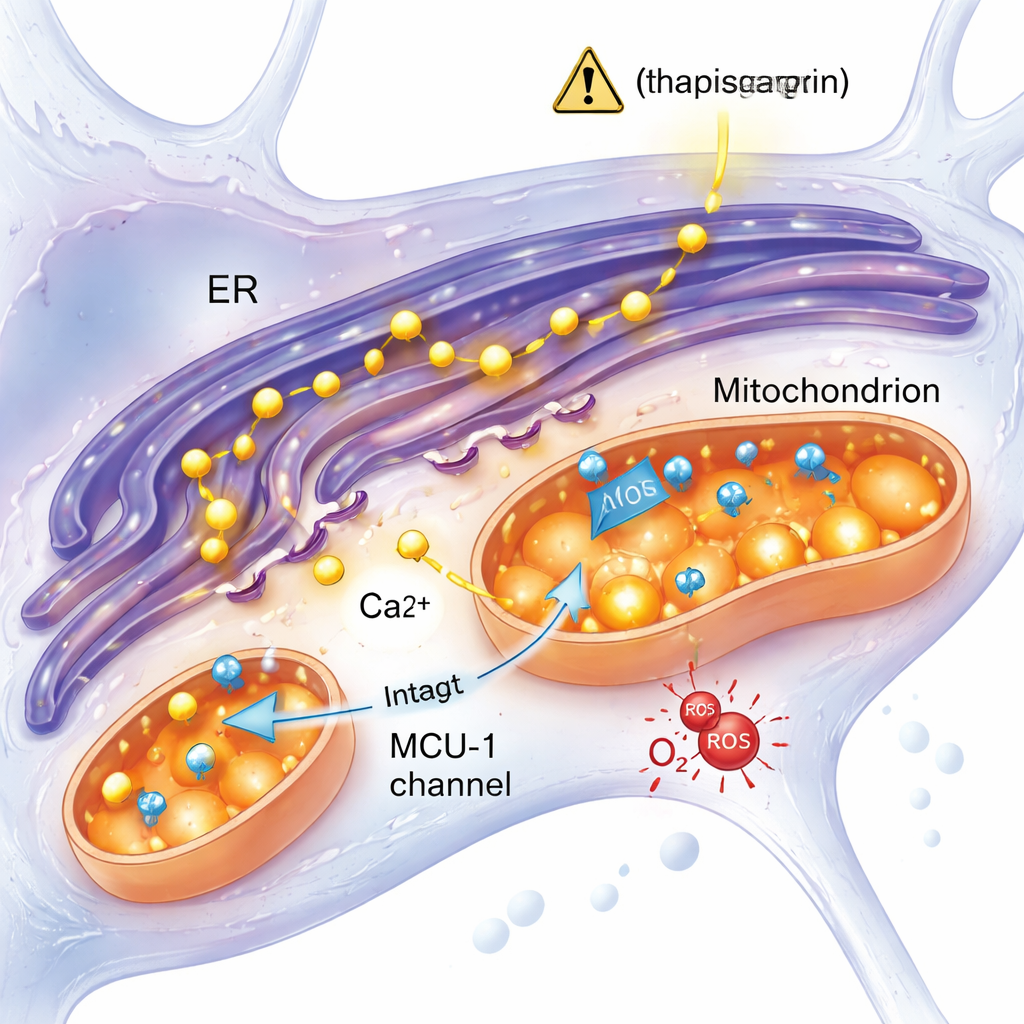
Eine Stressantwort unabhängig von einem wichtigen Calciumkanal
Eine führende Idee in der Alzheimer‑Forschung besagt, dass überschüssiges Calcium aus dem endoplasmatischen Retikulum durch einen Kanal namens mitochondrialer Calcium‑Uniporter (MCU‑1) in die Mitochondrien strömt, diese überlädt und Dysfunktion verursacht. Diese Studie prüfte diese Vorstellung direkt. Als das Team MCU‑1 mit einem spezifischen Inhibitor blockierte, zeigten sowohl Kontroll‑ als auch Mutanten‑Neuronen starke Reduktionen der mitochondrialen Calciumaufnahme, was bestätigte, dass der Kanal in beiden Gruppen funktionierte. Zudem reagierten Mutanten‑ und Kontrollzellen ähnlich, wenn die Calciumfreisetzung über einen physiologischeren Weg über den IP3‑Rezeptor ausgelöst wurde – ein weiterer wichtiger Calcium‑Mechanismus. Diese Ergebnisse sprechen gegen einen defekten MCU‑1‑Kanal und deuten stattdessen darauf hin, dass die physikalischen und funktionellen Kontakte zwischen endoplasmatischem Retikulum und Mitochondrien oder andere Aspekte ihrer Interaktion in den mutierten Neuronen verändert sind.
Was das für das Verständnis und die Behandlung der Krankheit bedeutet
Insgesamt zeichnen die Befunde das Bild von menschlichen Neuronen mit der PS1 M146L‑Alzheimer‑Mutation als Zellen, die im Ruhezustand normal erscheinen, aber unter Stress abnormal reagieren. Ihre Mitochondrien nehmen bei plötzlicher Freisetzung von internem Calcium nicht genügend Calcium auf, laufen jedoch heißer – sie verbrauchen mehr Sauerstoff und erzeugen mehr reaktive Sauerstoffspezies – als befänden sie sich in einem kostenintensiven Kompensationsmodus. Da dies in lebenden, menschlich abgeleiteten Neuronen geschieht, bevor klinische Symptome auftreten, stützt die Arbeit die Idee, dass gestörte Calcium‑Signale und frühe mitochondriale Überlastung upstream‑Ereignisse bei Alzheimer sind und nicht nur späte Nebenprodukte. Für Nicht‑Spezialisten lautet die Kernbotschaft: Das Gleichgewicht zwischen Calcium‑Signalen und mitochondrialer Energieproduktion aufrechtzuerhalten könnte genauso zentral für die Krankheitsprävention sein wie die Bekämpfung der bekannteren Amyloid‑Plaques.
Zitation: Wilson, C., Galeano, P., Remedi, M.M. et al. Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism. Sci Rep 16, 6002 (2026). https://doi.org/10.1038/s41598-026-35597-0
Schlüsselwörter: Alzheimer‑Krankheit, Mitochondrien, Calcium‑Signalisierung, Presenilin‑1‑Mutation, aus iPS‑Zellen abgeleitete Neuronen