Clear Sky Science · de
DFT‑Studie zu benzothiadiazol‑basierten kleinen Molekülen für hocheffiziente organische Photovoltaik
Warum bessere Solarmaterialien wichtig sind
Solarzellen werden auf Dächern und in Freiflächen immer häufiger, doch die zugrundeliegende Technologie entwickelt sich weiterhin rasant. Die derzeit effizientesten kommerziellen Module basieren auf starren Siliziumwafern: sie sind wirksam, aber teuer, schwer und nur schwer in gekrümmte Flächen oder leichte Geräte integrierbar. Diese Arbeit untersucht eine neue Klasse maßgeschneiderter organischer Moleküle, die dünnere, günstigere und flexiblere Solarzellen ermöglichen könnten — womit Fenster, Kleidung oder tragbare Geräte selbst zu Energiequellen werden könnten.
Von starren Modulen zu flexiblen Folien
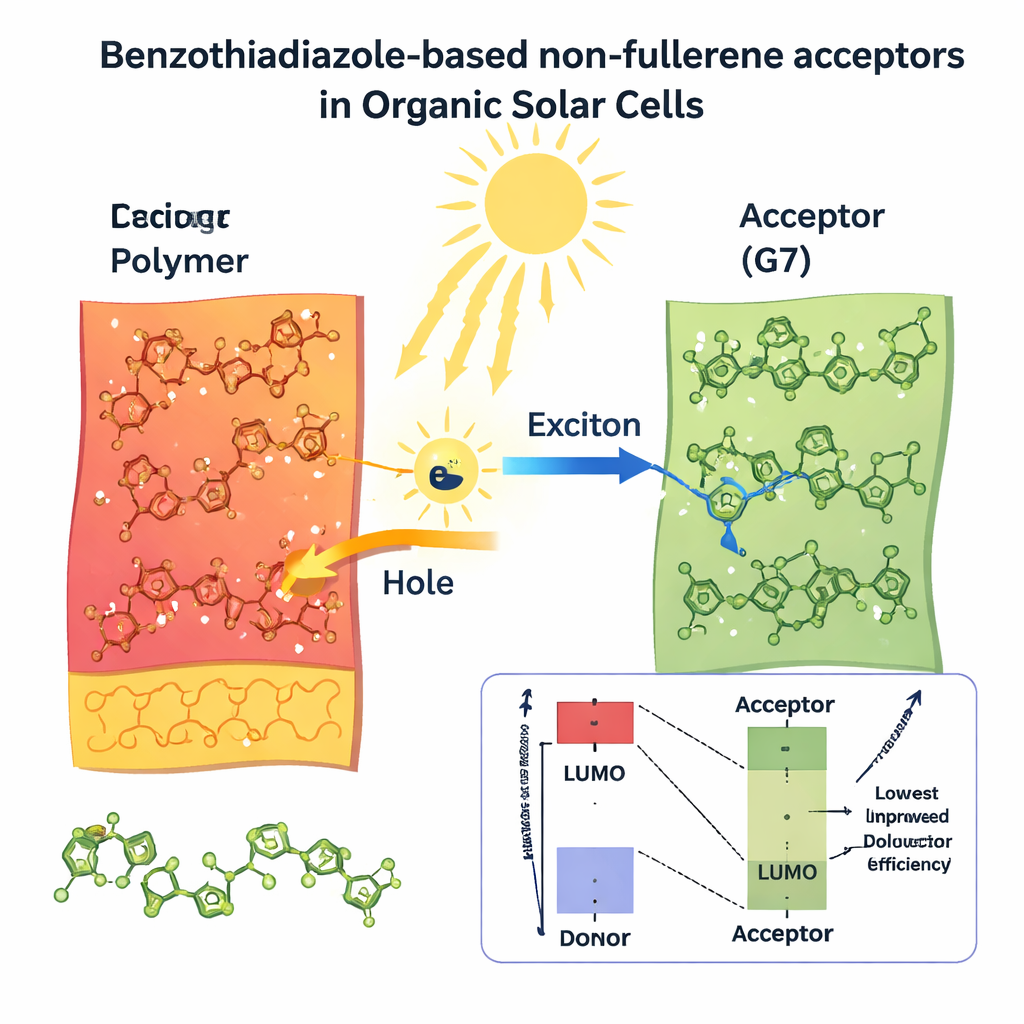
Konventionelle Silizium‑Solarzellen sind sehr effizient bei der Umwandlung von Sonnenlicht in Strom, bringen aber Kompromisse mit sich: Sie sind spröde, benötigen Hochtemperatur‑Fertigung und lassen sich nur schwer in leichte oder biegsame Produkte integrieren. Organische Solarzellen, aufgebaut aus kohlenstoffbasierten Molekülen, versprechen etwas anderes. Sie lassen sich wie Tinte drucken, chemisch feinabstimmen und als ultradünne Filme auf flexiblem Kunststoff aufbringen. Um ihr volles Potenzial auszuschöpfen, benötigen sie jedoch lichtabsorbierende Materialien, die mehr vom Sonnenspektrum einfangen und Ladungen mit minimalen Verlusten transportieren. Diese Studie konzentriert sich darauf, solche Materialien am Computer zu entwerfen, bevor sie im Labor synthetisiert werden.
Neue Bausteine am Bildschirm entwerfen
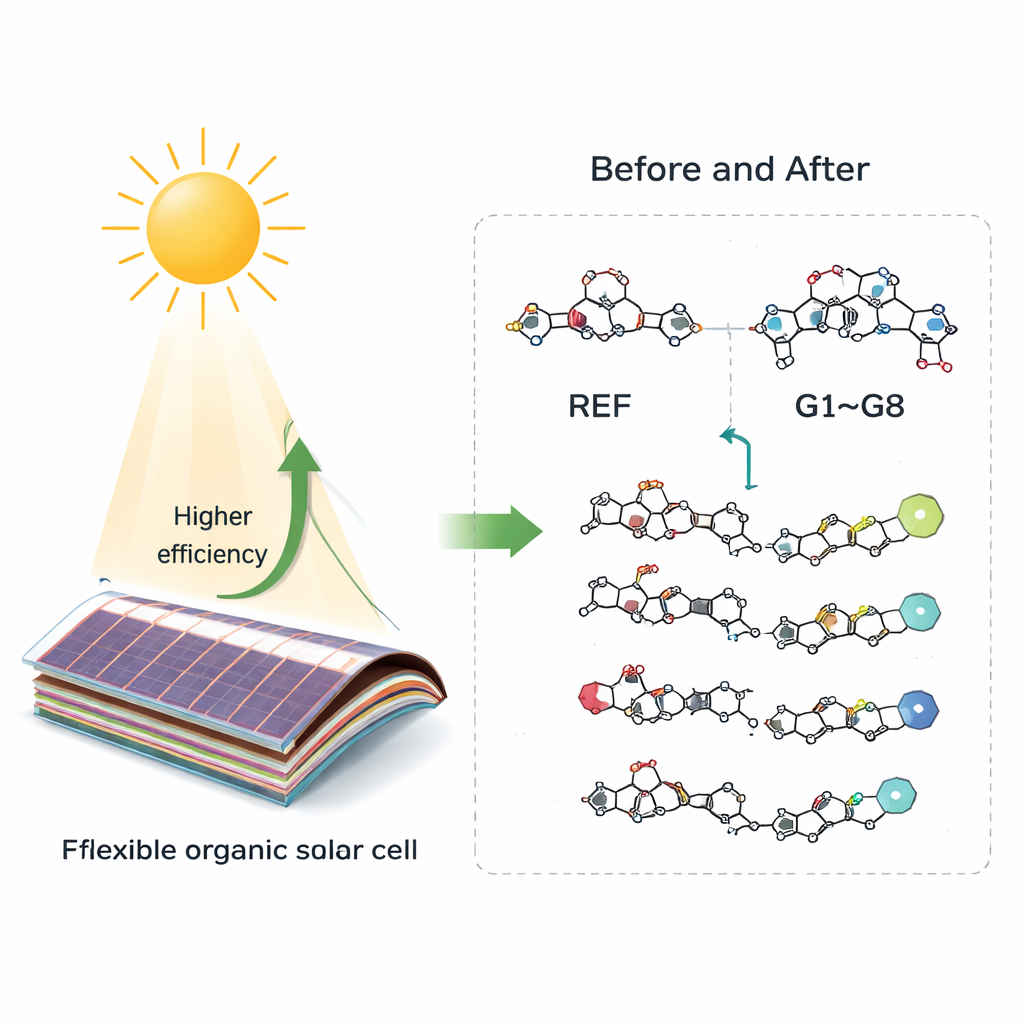
Die Forschenden begannen mit einem bekannten kleinen Molekül aus der organischen Elektronik und vereinfachten es zu einer Referenzstruktur, genannt REF. Diese Referenz dient als Rückgrat, bestehend aus einem zentralen «Donor»-Segment, flankiert von zwei «Akzeptor»-Segmenten. Das Team entwickelte dann acht neue Varianten (G1–G8), indem es die chemischen Gruppen an den Molekülenden austauschte. Diese terminalen Gruppen sind wie verstellbare Regler: Durch die Wahl stärkerer oder schwächerer elektronenziehender Enden lässt sich steuern, wie das Molekül Licht absorbiert und wie leicht es Ladungen weiterleitet. Mit quantenmechanischen Simulationen (einem Bereich der Theorie, bekannt als Dichtefunktionaltheorie) sagten sie die Absorptionsfarbe, die elektrischen Energieniveaus und die potenzielle Effizienz jedes Moleküls in einer Solarzelle voraus.
Mehr Sonnenlicht einfangen, weniger Energie vergeuden
Die virtuellen Experimente zeigten, dass alle acht neuen Entwürfe die ursprüngliche Rückgratstruktur in wichtigen Punkten übertreffen. Ihre Bandlücken — der Unterschied zwischen den Energieniveaus, auf denen sich Elektronen befinden, und denen, auf denen sie sich frei bewegen können — sind kleiner als bei REF, was bedeutet, dass sie röteres und nahes Infrarotlicht absorbieren, das Silizium und viele ältere organische Materialien ungenutzt lassen. Ein besonders auffälliger Kandidat, G7, absorbiert stark um 803 Nanometer, tief im roten Bereich, und erreicht in den Simulationen eine nahezu perfekte Lichtauffang‑Effizienz nahe 100 %. Mehrere Moleküle zeigen zudem sehr niedrige «Reorganisationsenergien», ein Maß dafür, wie stark sich die Molekülstruktur bei der Ladungsbewegung verformen muss. Niedrigere Werte bedeuten schnelleren, reibungsloseren Ladungstransport und geringere Verluste in einem funktionierenden Bauelement.
Spannung, Strom und Gesamtleistung ausbalancieren
Gute Solarmaterialien müssen mehr leisten als nur Licht zu absorbieren: Sie müssen hohe Spannungen erzeugen, starken Strom liefern und ohmsche Verluste gering halten. Die Autorinnen und Autoren schätzten diese praktischen Kennwerte — Leerlaufspannung, Füllfaktor und Gesamtstromwirkungsgrad — indem sie ihre Quantenberechnungen mit etablierten Geräte‑Modellen kombinierten. Sie prognostizieren, dass alle acht neuen Moleküle prinzipiell Wirkungsgrade über 20 % erreichen könnten, deutlich über den geschätzten 12 % für die ursprüngliche Referenzstruktur. Zwei Kandidaten heben sich aus unterschiedlichen Gründen hervor. G7 liefert den höchsten vorhergesagten Strom, da es den breitesten Anteil des Sonnenlichts einfängt und sich somit für Tandem‑ oder Niedriglichtanwendungen anbietet. G5 hingegen bietet das beste Gesamtgleichgewicht: Im Modell liefert es starken Strom, hohe Spannung und einen ausgezeichneten Füllfaktor, was zu einem projizierten Wirkungsgrad von etwa 37 % unter Standardsonnenlicht führt.
Was das für die zukünftige Solartechnik bedeutet
Für Nicht‑Expertinnen und Nicht‑Experten lautet die Kernaussage: Chemie kann wie ein Feineinstellrad für Solarmaterialien eingesetzt werden. Durch den Austausch nur der kleinen Gruppen an den Enden eines ansonsten ähnlichen Moleküls konnten die Forschenden große Verbesserungen in der Lichtaufnahme und der Umwandlungseffizienz vorhersagen. Obwohl diese Ergebnisse theoretisch sind und noch im Labor bestätigt werden müssen, weisen sie auf ein klares Rezept für das Design der nächsten Generation organischer Solarzellen hin: Terminaleinheiten so gestalten, dass sie die Lichtabsorption erweitern, eine saubere Ladungstrennung begünstigen und die molekulare Bewegung während des Ladungstransports minimieren. Unter den virtuellen Kandidaten sticht G7 durch seine Lichtauffangstärke hervor, während G5 die praktischste Allround‑Leistung bietet — beides starke Anwärter für künftige flexible, hocheffiziente Solarfolien.
Zitation: Ghaffar, A., Yousuf, A., Qureshi, M.Z. et al. DFT study of benzothiadiazole based small molecules for high efficiency organic photovoltaics. Sci Rep 16, 5859 (2026). https://doi.org/10.1038/s41598-026-35432-6
Schlüsselwörter: organische Solarzellen, nicht‑fullerene Akzeptoren, benzothiadiazol, photovoltaischer Wirkungsgrad, molekulares Design