Clear Sky Science · de
Untersuchung der biochemischen Grundlage der Resistenz gegen systemische Amyloidose
Wenn winzige Proteinveränderungen eine tödliche Anhäufung verhindern
Viele chronische Entzündungskrankheiten, von rheumatoider Arthritis bis Tuberkulose, können eine seltene, aber häufig tödliche Komplikation auslösen: die systemische AA-Amyloidose. Dabei lagert sich ein normales Blutprotein in Form starrer Fasern ab, die Organe verstopfen. Diese Studie stellt eine überraschend hoffnungsvolle Frage: Können kleine, natürliche Veränderungen dieses Proteins einige Tiere weitgehend immun gegen die Krankheit machen — und wenn ja, wie?
Die verborgene Gefahr von Proteinanhäufungen
Die AA-Amyloidose beginnt mit einem entzündlichen Alarmzeichen im Blut, dem Serum-Amyloid A (SAA). Bei starker oder lang andauernder Entzündung kann der SAA-Spiegel um das Tausendfache ansteigen. Bei manchen Menschen und Tieren faltet sich ein Teil dieses Proteins fehl und lagert sich zu langen Fasern zusammen, sogenannten Amyloidfibrillen, die sich in Organen wie Milz und Nieren ausbreiten. Mit der Zeit beeinträchtigen diese Fasern die Organfunktion. Doch nicht jeder mit hohem SAA-Spiegel entwickelt Amyloidose, und bestimmte Mausstämme bleiben selbst bei experimenteller Anfälligkeit überraschend resistent. Zu verstehen, warum das so ist, könnte neue Strategien zur Verhinderung von Amyloidablagerungen beim Menschen aufzeigen.
Resistente Mäuse und ihre besonderen Proteinvarianten
Bei Mäusen entstehen die meisten AA-Amyloidfibrillen aus einer Variante von SAA namens SAA1.1, die stark mit der Krankheit verbunden ist. Einige Mausstämme produzieren jedoch überwiegend leicht veränderte Varianten, SAA1.5 und SAA2.2, und diese Stämme entwickeln kaum systemische AA-Amyloidose. Die Proteine unterscheiden sich nur in wenigen Bausteinen (Aminosäuren), doch diese Unterschiede konzentrieren sich in einer dicht gepackten Region, die den inneren Kern der krankheitsverursachenden Fasern bildet. Die Forscher schlugen vor, dass diese kleinen Unterschiede das Protein nicht vollständig am Aggregieren hindern, sondern vielmehr verhindern, dass es genau jene spezifische, schädliche Faserstruktur annimmt.
Die Proteine im Labor auf die Probe gestellt
Um diese Idee zu prüfen, stellten die Autoren alle drei Maus-SAA-Varianten in Bakterien her und beobachteten ihr Verhalten in Reagenzglas-Experimenten. Sie verfolgten die Faserbildung mit einem fluoreszierenden Farbstoff, der aufleuchtet, wenn Amyloid entsteht, und bestätigten Strukturen mittels Elektronenmikroskopie. Das krankheitsassoziierte SAA1.1 bildete leicht lange, gerade Fasern. SAA2.2 konnte ebenfalls Fasern bilden, diese waren jedoch dicker, stärker gedreht und strukturell variabler und lösten nicht das gleiche starke Farbstoffsignal aus. SAA1.5 dagegen bildete unter den getesteten Bedingungen keine Fasern. Als die Wissenschaftler kleine Proben echter Krankheitsfasern aus kranken Maussplen als "Samen" hinzufügten, wuchs SAA1.1 schnell zu neuen Fasern heran, die die Struktur des Originals eng kopierten, ähnlich einem Prion. Auffällig war, dass SAA1.5 und SAA2.2 überhaupt nicht auf diese Samen aufwuchsen; die ex vivo gewonnenen Fasern konnten sie nicht in die pathogene Form einreihen.
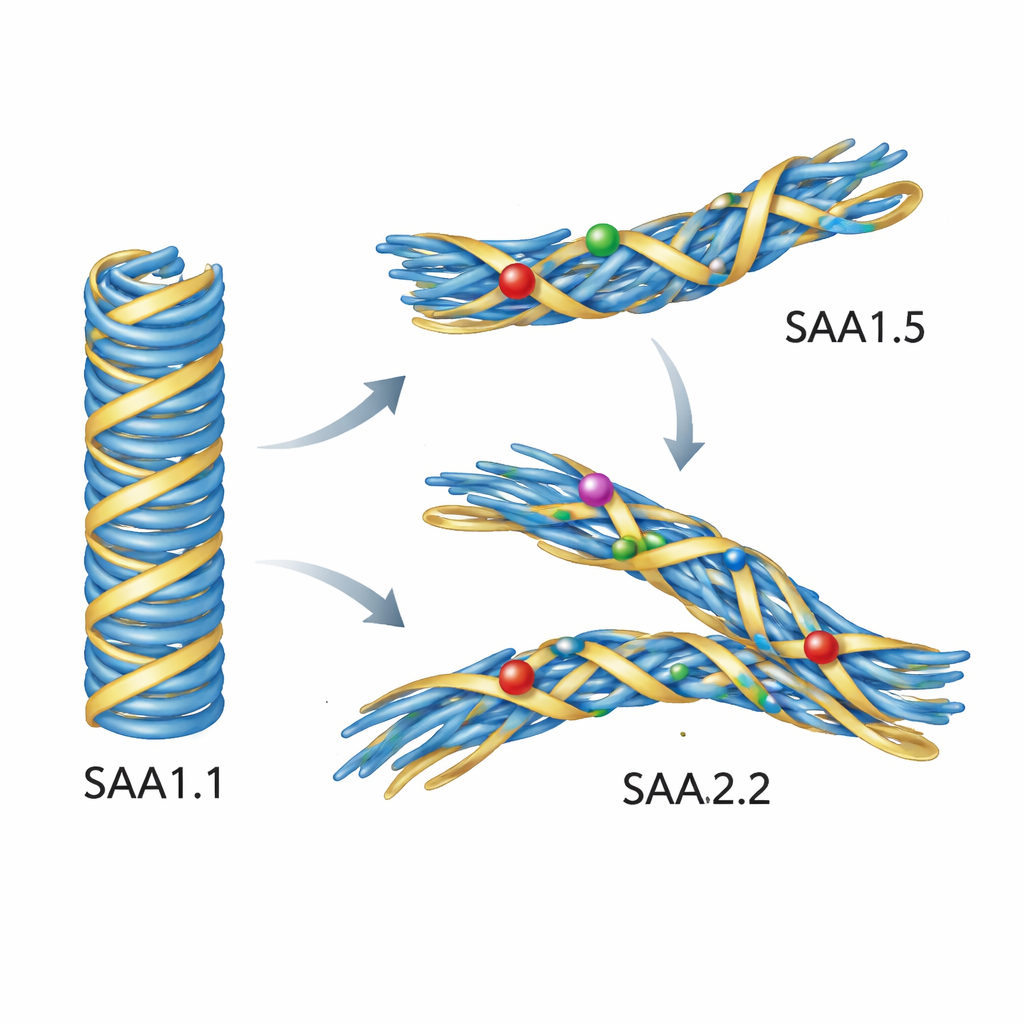
Simulationen zeigen, warum mutierte Proteine die schädliche Form ablehnen
Experimentelle Befunde allein konnten nicht auf atomarer Ebene zeigen, was genau schiefgeht, deshalb wandten sich die Autoren detaillierten Computersimulationen zu. Sie begannen mit einer hochauflösenden Struktur einer pathogenen Maus-AA-Fibrille aus SAA1.1 und setzten dann rechnerisch die Sequenzen von SAA1.5 und SAA2.2 ein. In Simulationen dieser Fasern in Wasser bei Körpertemperatur blieb das auf SAA1.1 basierende Modell bemerkenswert stabil. Dagegen verschoben und verzerrten sich Fasern, die aus SAA1.5 oder SAA2.2 aufgebaut waren. Eine Schlüssel-Loop-Region im Kern bewegte sich nach außen und löste Kontakte mit dem N-terminalen Segment des Proteins, und mehrere Seitenketten drehten sich in neue Orientierungen. Diese subtilen Umordnungen störten die enge Packung, die die krankheitsassoziierte Faltstruktur definiert. Anders gesagt: Die Varianten sequenzen hatten grundsätzlich nichts dagegen, Fasern zu bilden — aber sie passten nicht bequem in die Blaupause der pathogenen AA-Fibrille.
Wie die Designprinzipien der Natur auf künftige Therapien hinweisen
Insgesamt zeigen die Ergebnisse, dass "amyloidresistente" Mausstämme nicht geschützt sind, weil ihr SAA sich überhaupt nicht aggregieren kann. Vielmehr sind ihre SAA-Varianten strukturell inkompatibel mit genau jener Faserform, die die systemische AA-Amyloidose verursacht. Die Proteine können weiterhin verklumpen, tun dies aber in alternativen, offenbar harmlosen Formen. Ähnliche schützende Mutationen sind aus anderen Proteinfehlfaltungs-Erkrankungen bekannt, einschließlich einiger Prion- und Alzheimer-Fälle. Das deutet auf ein allgemeineres Prinzip hin: Eine Krankheit begünstigende Proteinfaltung so zu verändern, dass das toxische Architekturmuster nicht mehr eingenommen werden kann — während die normale Funktion erhalten bleibt — könnte ausreichen, um Krankheit zu verhindern. Langfristig könnten von diesen natürlichen "resistenten" Varianten oder von kurzen Fragmenten, die sich von ihnen ableiten, inspirierte Therapien dazu beitragen, Proteine von schädlichen Faltungen weg und zu harmloseren Formen hinzulenken.
Zitation: Moderer, T., Schnell, A.F., Scheurmann, N.J. et al. Assessment of the biochemical basis underlying the resistance against systemic amyloidosis. Sci Rep 16, 1313 (2026). https://doi.org/10.1038/s41598-026-35297-9
Schlüsselwörter: AA-Amyloidose, Serum-Amyloid A, Proteinfehlfaltung, Amyloidresistenz, Mausmodelle