Clear Sky Science · de
Stochastisches LASSO für extrem hochdimensionale genomische Daten
Die Nadeln im genomischen Heuhaufen finden
Die moderne Biologie kann Zehntausende Gene gleichzeitig messen, doch Studien mit Patientinnen und Patienten umfassen oft nur wenige hundert Personen. In diesem Ungleichgewicht verbergen sich kleine Genmengen, die tatsächlich wichtig sind, um Krankheitsrisiko oder Überleben vorherzusagen. Dieses Papier stellt „Stochastic LASSO“ vor, eine statistische Methode, die darauf ausgelegt ist, diese Schlüsselgene zuverlässig aus Ozeanen verrauschter genomischer Daten zu entdecken — selbst wenn es deutlich mehr Gene als Probanden gibt.
Warum die richtige Genwahl so schwierig ist
Forscher nutzen häufig Werkzeuge wie LASSO, die unbedeutende Geneffekte gegen null schrumpfen, während die informativsten Effekte erhalten bleiben. Klassische LASSO‑Varianten geraten jedoch an ihre Grenzen, wenn die Zahl der Gene die Zahl der Proben weit übersteigt, was in der Krebsgenomik üblich ist. Standard‑LASSO kann höchstens so viele Gene auswählen wie es Patienten gibt, und es übersieht tendenziell Gene mit ähnlichem Verhalten. Frühere Verbesserungen, die zusätzliche Bestrafungsbegriffe einführen, können mit einem Teil dieser Korrelation umgehen, verwischen dabei aber möglicherweise die biologische Bedeutung, indem verwandte Gene so behandelt werden, als würden sie alle das Ergebnis in die gleiche Richtung beeinflussen.
Bessere zufällige Stichproben erzeugen
Ein vielversprechender Workaround besteht darin, LASSO wiederholt auf vielen kleineren, zufällig gezogenen Genuntermengen anzuwenden und die Ergebnisse zu kombinieren. Diese „Bootstrap“‑Ansätze leiden jedoch weiterhin an drei Problemen: korrelierte Gene können sich gegenseitig auslöschen, viele Gene werden selten oder nie gezogen, und reine Zufälligkeit macht die endgültige Auswahl instabil. Stochastic LASSO geht diese Probleme direkt an mit einem neuen Stichprobenverfahren namens korrelationsbasierte Bootstrap‑Stichprobe. Anstatt Gene rein zufällig auszuwählen, bevorzugt es gezielt Gene, die weniger mit bereits gewählten Genen korreliert sind, und erzeugt so kleinere Genmengen, die deutlich unabhängiger sind. Zudem stellt es sicher, dass jedes Gen über die Bootstrap‑Durchläufe hinweg gleich oft verwendet wird, sodass kein Gen ungerechtfertigt ignoriert wird. 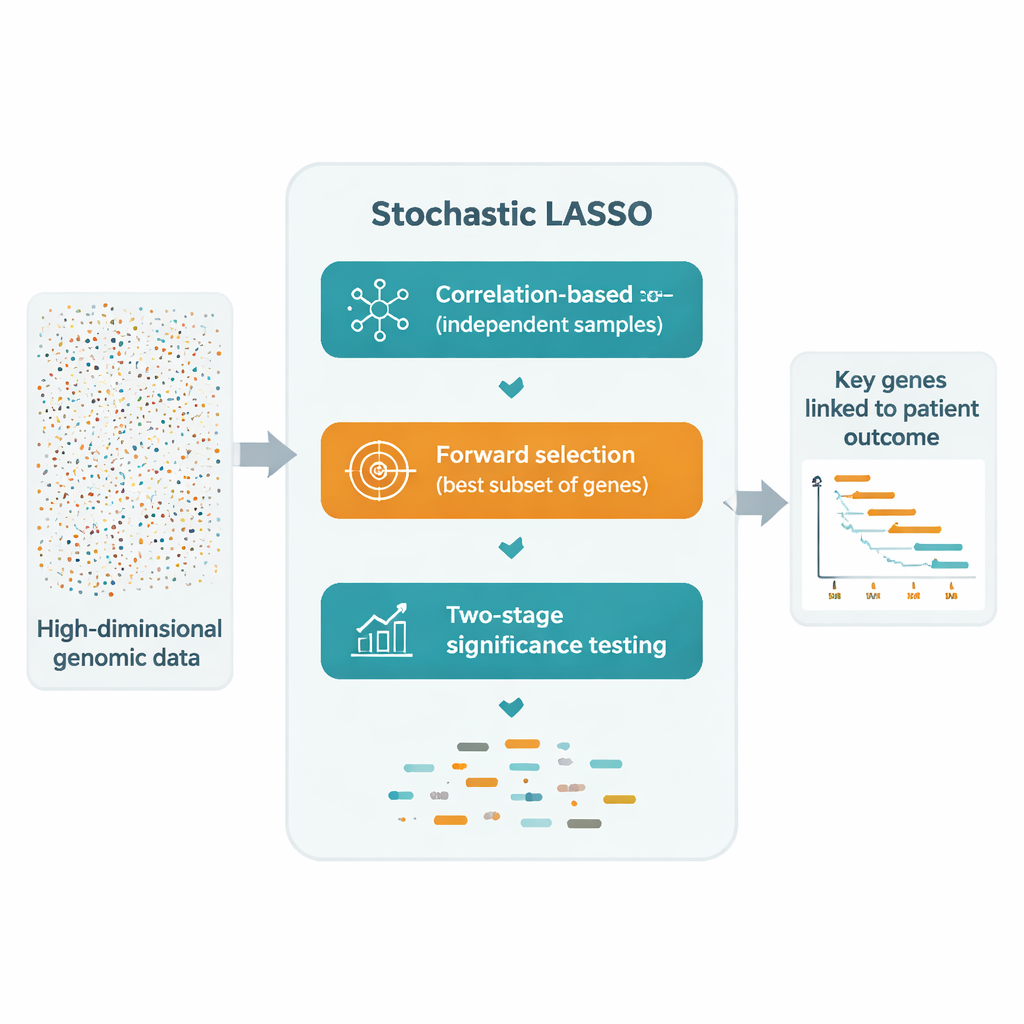
Von lokalen Hinweisen zu einer globalen Genmenge
Nachdem diese saubereren Untergruppen erstellt sind, zeichnet Stochastic LASSO auf, wie groß der Koeffizient jedes Gens über alle Bootstrap‑Fits hinweg ist. Dieser durchschnittliche absolute Effekt wird zu einem „lokalen Score“, der widerspiegelt, wie konsistent wichtig das Gen erscheint. Anstatt jede mögliche Kombination erschöpfend zu testen, baut die Methode Kandidatenmodelle auf, indem Gene in der Reihenfolge ihrer lokalen Scores hinzugefügt werden, und bewertet, wie gut jedes Kandidatmodell Vorhersagen an separaten Validierungsdaten liefert. Auf diese Weise wählt sie eine kompakte Genmenge aus, deren kombinierte Signale die Daten am besten erklären — und das mit weit weniger Durchläufen als traditionelle schrittweise Verfahren.
Testen, welche Gene wirklich wichtig sind
Um von „oft ausgewählt“ zu „statistisch überzeugend“ zu gelangen, führen die Autoren einen zweistufigen t‑Test ein. Zuerst prüfen sie, ob sich der mittlere Koeffizient jedes Gens über die Bootstraps klar von null unterscheidet und markieren es als potenziell bedeutsam. Unter diesen Kandidaten fragen sie dann, ob der Effekt eines Gens größer ist als die typische Effektgröße aller Kandidaten. Nur Gene, die beide Tests bestehen, werden als signifikant erklärt. Da diese Tests auf vielen Bootstrap‑Schätzungen beruhen, kann Stochastic LASSO mit Zuversicht mehr signifikante Gene identifizieren, als es Patienten gibt — etwas, das konventionelles LASSO nicht leisten kann. 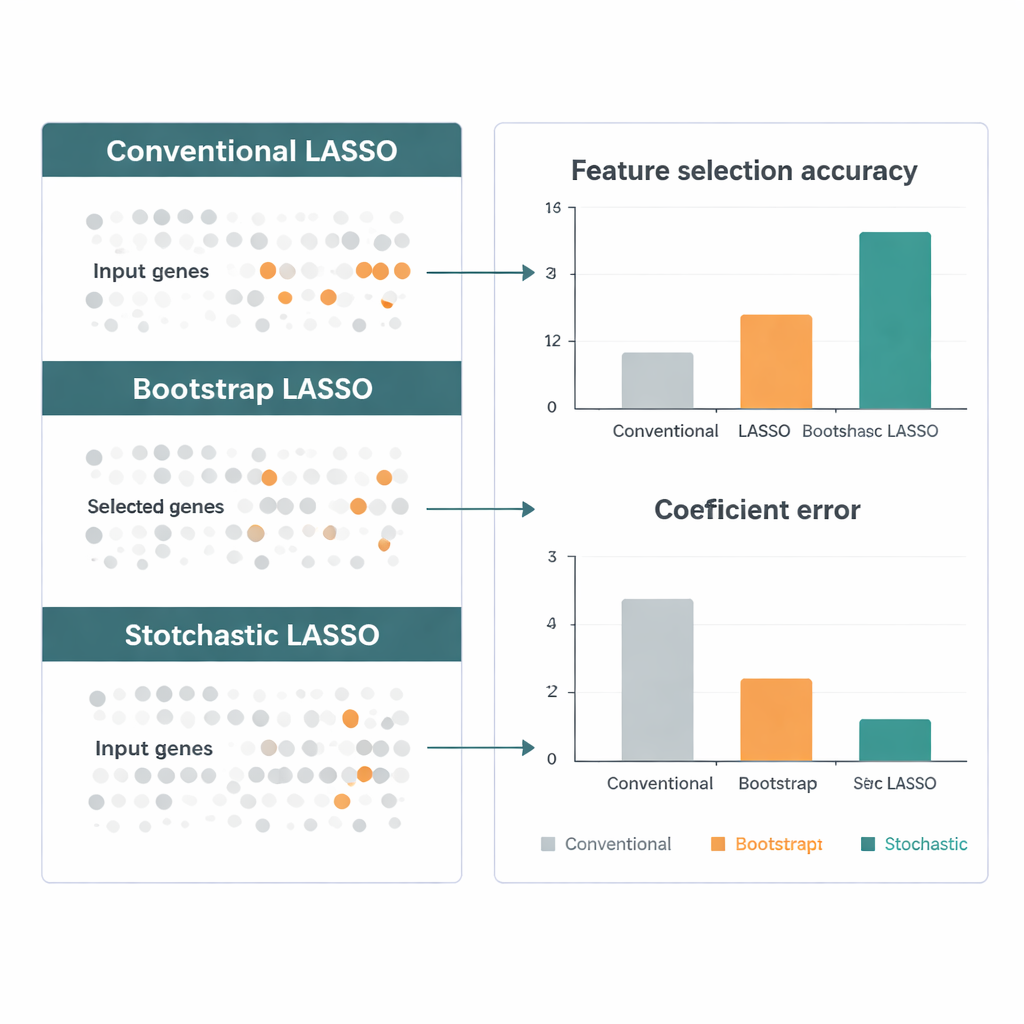
Seinen Wert in Simulationen und Krebsdaten beweisen
Die Autoren vergleichen Stochastic LASSO mit mehreren führenden LASSO‑Varianten anhand simulierten Daten, die reale genomische Studien nachbilden sollen: sehr viele Gene, starke Korrelationen und bekannte „wahre“ Signale. In zahlreichen Szenarien findet die neue Methode die richtigen Gene häufiger, schätzt deren Effekte genauer und bleibt von Lauf zu Lauf stabil. Anschließend wenden sie sie auf Genexpressionsdaten aus The Cancer Genome Atlas für Hirntumoren an, einschließlich aggressivem Glioblastom. Stochastic LASSO hebt Hunderte von Genen hervor, deren Aktivität mit dem Überleben der Patientinnen und Patienten zusammenhängt, und weist auf biologische Signalwege — etwa Signaltransduktion und Wirkstoff‑Metabolisierungswege — hin, die in der Literatur unabhängig gestützt werden, was darauf hindeutet, dass die Methode nicht nur statistisch schärfer, sondern auch biologisch sinnvoll ist.
Was das für Patientinnen, Patienten und Forschende bedeutet
Für Nicht‑Spezialisten lautet die Kernbotschaft: Stochastic LASSO ist ein intelligenterer Filter für genomische Big Data. Er hilft Forschenden, echte krankheitsbezogene Gene von statistischem Rauschen zu trennen, selbst wenn die Daten begrenzt sind und Gene stark miteinander verknüpft sind. Indem er genauere und stabilere Genlisten sowie Effekt‑Schätzungen liefert, kann er die Suche nach Biomarkern, Wirkstoffzielen und prognostischen Signaturen bei Krebs und anderen komplexen Erkrankungen schärfen. Obwohl die Demonstration auf linearer Regression basiert, lässt sich derselbe Rahmen in Überlebensmodelle und Klassifikationsprobleme einbetten und erweitert so das mögliche Wirkungsspektrum in der biomedizinischen Forschung.
Zitation: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
Schlüsselwörter: Auswahl genomischer Merkmale, hochdimensionale Daten, LASSO‑Methoden, Genexpressionsdaten bei Krebs, Biomarker‑Entdeckung