Clear Sky Science · de
Lactat reguliert die YTHDF2-FTH1-Achse, um Ferroptose von Kardiomyozyten zu fördern und die ischämisch-reperfusionsbedingte Myokardschädigung zu verschlimmern
Warum Herzpatienten diese Chemie interessieren sollte
Wenn Ärzte nach einem Herzinfarkt eine verstopfte Herzarterie wiedereröffnen, rettet der Zustrom frischen Bluts Muskelgewebe, kann aber auch zusätzlichen Schaden verursachen – die sogenannte Ischämie‑Reperfusions‑Schädigung. Diese Studie legt einen überraschenden Schuldigen innerhalb der Herzmuskelzellen offen: das verbreitete Stoffwechselzwischenprodukt Lactat. Die Autorinnen und Autoren zeigen, dass Lactat einen molekularen Schalter umlegt, der Herzzellen in Richtung einer bestimmten Form des eisenabhängigen Zelltods treibt und so die Schädigung verschlimmert. Das Verständnis dieses verborgenen Weges könnte auf neue Medikamente hinweisen, die das Herz während der Notfallbehandlung besser schützen.
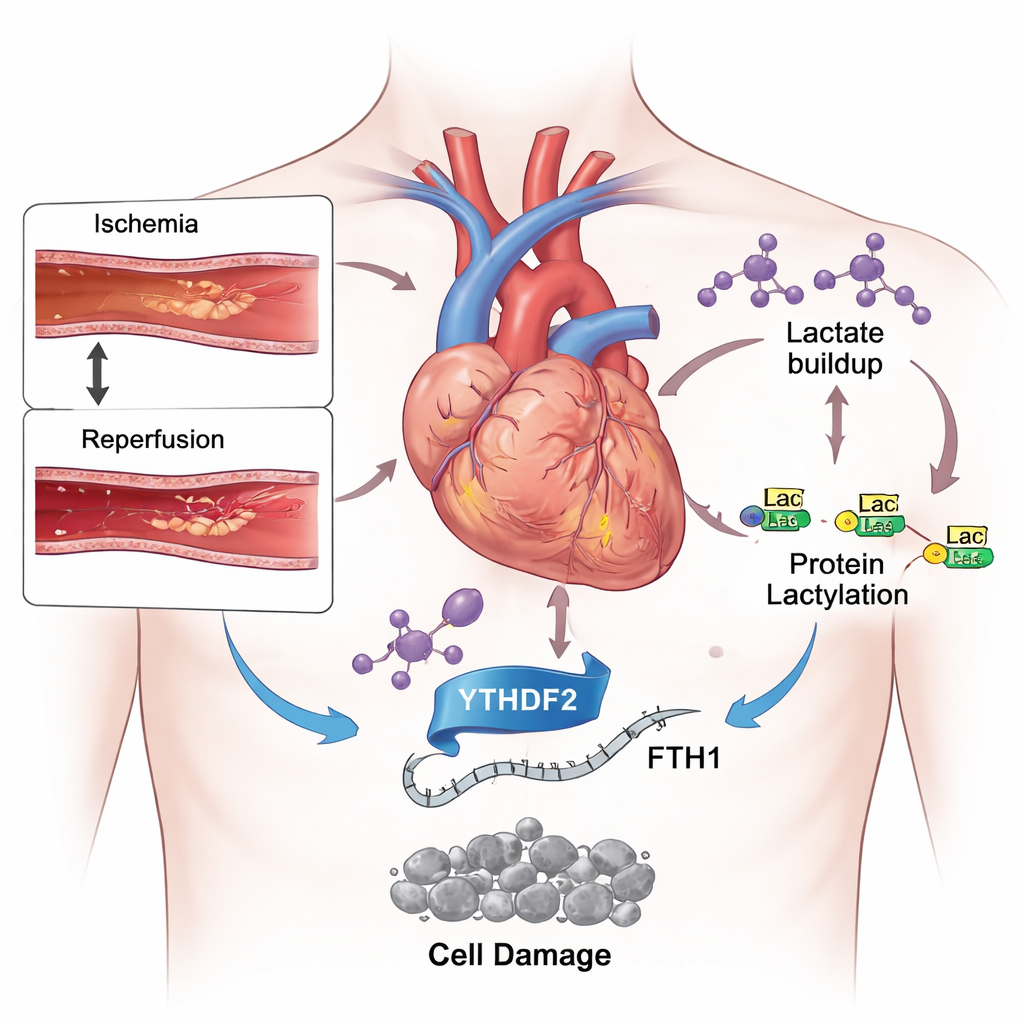
Ein zweischneidiges Schwert bei der Behandlung von Herzinfarkten
Die moderne Medizin ist sehr gut darin geworden, verstopfte Koronararterien schnell wieder zu öffnen und so den unmittelbaren Schaden eines Herzinfarkts zu begrenzen. Dennoch verlieren Patienten nach Wiederherstellung des Blutflusses weiterhin große Bereiche des Herzmuskels. Ein Grund ist, dass die plötzliche Rückkehr von Sauerstoff und Nährstoffen einen Sturm chemischen Stresses in den Herzmuskelzellen auslöst. Unter mehreren in diesem Kontext ausgelösten Zelltodtypen hat eine neuere Form, die Ferroptose, Aufmerksamkeit erregt. Im Gegensatz zu bekannteren Formen wie der Apoptose hängt Ferroptose von Eisen und einer enthemmten Oxidation von Fetten in Zellmembranen ab, was das Herz dauerhaft schwächen kann.
Wie Lactat mehr wird als nur „Muskelbrennen“
Während eines Herzinfarkts verlagert der unterversorgte Herzmuskel seinen Energieverbrauch zugunsten der Glykolyse, eines Backup‑Systems, das Zucker schnell abbaut, dabei aber große Mengen Lactat erzeugt. Anhand von Mäusen, die einer kurzzeitigen Verlegung und Wiedereröffnung einer Herzarterie unterzogen wurden, sowie kultivierten herzähnlichen Zellen, die zunächst Sauerstoffmangel und anschließend Reoxygenierung ausgesetzt waren, fanden die Forschenden stark erhöhte Lactatspiegel. Gleichzeitig entdeckten sie mehr einer chemischen Markierung namens Lactylierung an vielen Proteinen und an Histonen, den Gerüsten, die die DNA organisieren. Gaben sie den Tieren ein Medikament, das die Glykolyse verlangsamt und die Lactatproduktion senkt, nahm der Herzschaden ab, Blutmarker für die Schädigung fielen, und das Gleichgewicht zwischen schädlichem Eisen und schützenden Antioxidantien verbesserte sich. Diese Ergebnisse legen nahe, dass überschüssiges Lactat nicht nur ein Stressnebenprodukt ist, sondern ein aktiver Treiber der Schädigung.
Ein molekularer Schalter, der die Leine des Eisens löst
Bei tieferer Untersuchung richtete das Team sein Augenmerk auf YTHDF2, ein Protein, das chemische Markierungen auf RNA liest und entscheidet, wie schnell bestimmte Botschaften abgebaut werden. Sie entdeckten, dass Ischämie‑Reperfusion und zugeführtes Lactat beide die YTHDF2‑Spiegel erhöhten und die Lactylierung rund um das Gen, das es kodiert, stärkten, wodurch seine Produktion verstärkt wurde. Eines der Hauptziele von YTHDF2 erwies sich als die RNA für die schwere Untereinheit der Ferritin‑Kette 1 (FTH1), ein zentraler Bestandteil des zellulären Eisenspeicherkäfigs. FTH1 verpackt Eisen normalerweise in einer sicheren Form, die verhindert, dass es schädliche Reaktionen antreibt. In gestressten Herzzellen band YTHDF2 stärker an die FTH1‑RNA und beschleunigte deren Abbau, sodass Zellen weniger Ferritinkäfige hatten, mehr freies Eisen vorhanden war, der oxidative Stress zunahm und typische Merkmale der Ferroptose auftraten.
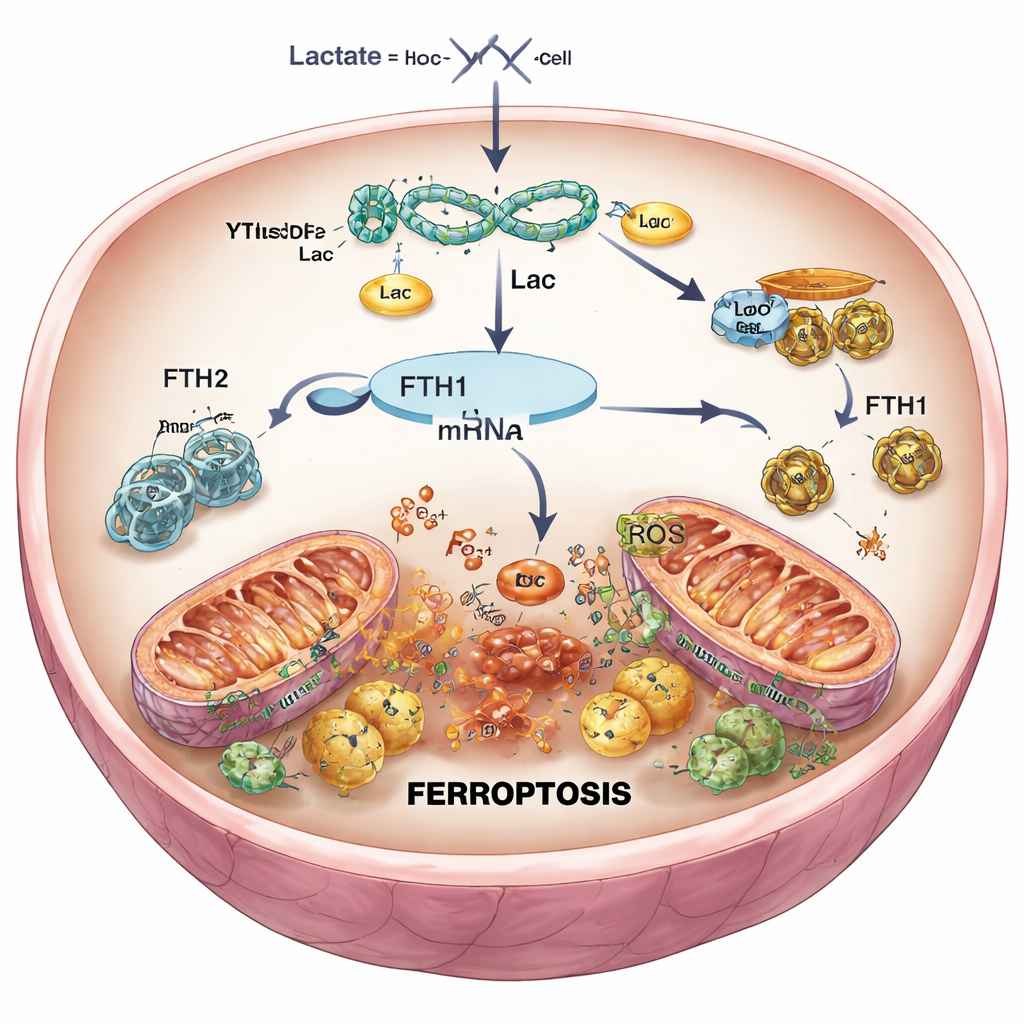
Das Totsignal in Herzzellen abschwächen
Um Ursache und Wirkung zu prüfen, nutzten die Forschenden genetische Werkzeuge, um YTHDF2 gezielt in Herzzellen und in Mäusen zu reduzieren. Wurde YTHDF2 herunterreguliert, erholten sich die FTH1‑Spiegel, Eisen und reaktive Sauerstoffspezies sanken, die Mitochondrien behielten eine eher normale Gestalt, und das Überleben der Zellen nach simulierten Reperfusionsbedingungen verbesserte sich insgesamt. Bei Mäusen führten geringere YTHDF2‑Spiegel zu kleineren Infarktnarben und gesünder wirkendem Gewebe. Wenn jedoch FTH1 gleichzeitig reduziert wurde, verschwanden diese Vorteile größtenteils: Das Eisen stieg wieder, oxidative Schäden kehrten zurück und die Infarktgröße nahm zu. Das bestätigte, dass YTHDF2 Ferroptose hauptsächlich fördert, indem es FTH1 unterdrückt und so die Kontrolle über das intrazelluläre Eisen lockert.
Was das für künftige Herztherapien bedeutet
Setzt man die Einzelteile zusammen, skizziert die Studie eine neue Ereigniskette: Eine blockierte und dann wieder eröffnete Arterie führt zu Lactatanhäufung; Lactat erhöht YTHDF2 durch Lactylierung; YTHDF2 zerstört daraufhin die RNA‑Anweisungen für das eisenbewahrende Protein FTH1; und die daraus resultierende Eisenüberladung löst Ferroptose aus und vertieft die Herzschädigung. Für Patientinnen und Patienten ist die Botschaft hoffnungsvoll: Dieser Weg bietet mehrere neue Ansatzpunkte für Interventionen. Medikamente, die schädliche Lactat‑Signale begrenzen, die spezifische Modifikation von YTHDF2 blockieren oder die FTH1‑Funktion erhalten, könnten die Notfall‑Reperfusion sicherer machen und mehr Herzmuskel schützen. Zwar müssen diese Befunde noch in menschlichem Gewebe bestätigt werden, doch sie eröffnen einen vielversprechenden Pfad zu schonenderen und wirksameren Behandlungen für Überlebende von Herzinfarkten.
Zitation: Xiang, Z., Xiang, B., Ouyang, T. et al. Lactate regulates the YTHDF2-FTH1 axis to promote cardiomyocyte ferroptosis and aggravate myocardial ischemia-reperfusion injury. Sci Rep 16, 4865 (2026). https://doi.org/10.1038/s41598-026-35130-3
Schlüsselwörter: Herzinfarkt, Lactat, eisenabhängiger Zelltod, Ischämie-Reperfusions-Schaden, Schutz von Kardiomyozyten