Clear Sky Science · de
DrugBank‑Analyse mit maschinellem Lernen enthüllt neue Kandidaten zur BCL‑2‑Hemmung
Warum schlauere Krebsmedikamente wichtig sind
Krebszellen weigern sich häufig zu sterben, wenn sie es sollten. Viele Tumoren übernutzen eine Familie von „Bodyguard“-Proteinen namens BCL‑2, die das eingebaute Selbstmordprogramm der Zelle blockieren. Es gibt bereits Wirkstoffe, die BCL‑2 ansprechen, doch sie können Nebenwirkungen verursachen und wirken nicht bei allen Patienten. Diese Studie untersucht, wie modernes maschinelles Lernen Tausende bereits existierender Medikamente durchsieben kann, um neue, sicherere Kandidaten zu finden, die BCL‑2 außer Kraft setzen und Krebszellen zur Selbstzerstörung bringen könnten.
Wie Zellen zwischen Leben und Tod wählen
Gesundes Gewebe entfernt kontinuierlich geschädigte oder nicht benötigte Zellen durch einen kontrollierten Selbstzerstörungsprozess, die Apoptose bzw. programmierter Zelltod. Eine Gruppe von Proteinen, die BCL‑2‑Familie, wirkt dabei als zentraler Schalter für diese Entscheidung. Einige Mitglieder fördern das Überleben der Zelle, andere treiben sie in den Tod. In vielen Krebsarten werden die Überlebens fördernden Mitglieder, darunter BCL‑2 und sein naher Verwandter BCL‑XL, im Überschuss produziert. Dieser zusätzliche Schutz erlaubt es Krebszellen, Todes signale zu ignorieren und Chemotherapien zu widerstehen. Deshalb ist die Blockade von BCL‑2 zu einer attraktiven Strategie in der Krebstherapie geworden, doch aktuelle Wirkstoffe treffen oft verwandte Proteine mit und führen so zu Nebenwirkungen wie gefährlichen Abfällen der Thrombozytenzahl.
Computern beibringen, vielversprechende Moleküle zu erkennen
Anstatt neue Verbindungen von Grund auf neu zu suchen, griffen die Forschenden auf Datenbanken von Molekülen zurück, die bereits untersucht oder als Medikamente eingesetzt wurden. Sie begannen mit einer großen öffentlichen Ressource namens ChEMBL, die experimentelle Messungen enthält, wie stark verschiedene Chemikalien an BCL‑2 binden. Nach sorgfältiger Bereinigung dieser Informationen — Entfernen von Duplikaten, unsicheren Messungen und ungewöhnlich großen oder seltenen Molekülen — blieben 601 gut charakterisierte Verbindungen übrig. Jedes Molekül wurde in eine Art digitalen Fingerabdruck übersetzt, der seine strukturellen Merkmale erfasst. Diese Fingerabdrücke wurden verwendet, um sieben verschiedene Modelle des maschinellen Lernens zu trainieren und zu vergleichen mit der Aufgabe, zu entscheiden, ob ein neues Molekül wahrscheinlich ein starker BCL‑2‑Blocker oder im Wesentlichen inaktiv ist. 
Das beste Modell auswählen und eine Arzneimittelbibliothek durchsuchen
Das Team bewertete die Modelle anhand eines separaten Testdatensatzes, der während des Trainings nicht verwendet worden war, und prüfte dabei nicht nur, wie oft jedes Modell richtig lag, sondern auch, wie gut es Aktive von Inaktiven unterschied und wie ausgewogen seine Vorhersagen waren. Ein Modell namens LightGBM — eine moderne, baumbasierte Boosting‑Methode — lieferte in den meisten Maßen die beste Leistung, einschließlich Gesamtgenauigkeit und der Fähigkeit, zuverlässige Wahrscheinlichkeiten zuzuweisen. Mit diesem feinabgestimmten Modell wandten sich die Forschenden DrugBank zu, einer kuratierten Sammlung von mehr als 12.000 zugelassenen, experimentellen und zurückgezogenen Arzneistoffen. Nachdem sie dieselben Fingerabdrücke berechnet hatten, fragten sie LightGBM, welche dieser Moleküle wie potenzielle BCL‑2‑Inhibitoren aussehen. Nur neun Verbindungen erhielten hohe Werte, etwa das oberste ein Zehntel von einem Prozent der gesamten Bibliothek, was zeigt, dass das virtuelle Screening sehr selektiv war. Vier der neun waren bereits bekannte BCL‑2‑Inhibitoren, was das Team beruhigte, dass der Ansatz sinnvoll war.
Von Computer‑Treffern zu molekularen Wechselwirkungen
Unter den verbleibenden hoch bewerteten Molekülen konzentrierten sich die Forschenden auf drei, die zuvor nicht mit BCL‑2 in Verbindung gebracht worden waren: Dersalazine, Opelconazole und Zongertinib. Um zu prüfen, ob diese Kandidaten plausibel in die Bindungstasche von BCL‑2 passen könnten, nutzten sie Computer‑Docking, eine Technik, die vorhersagt, wie ein kleines Molekül sich an die Oberfläche eines Proteins anschmiegt. Die Simulationen deuteten darauf hin, dass insbesondere Opelconazole und Zongertinib Netzwerke günstiger Kontakte mit denselben Schlüsselaminosäuren ausbilden, die ein gut untersuchtes Referenzarzneimittel, ABT‑737, greifen. Ihre vorhergesagten Bindungsstärken lagen nahe bei denen etablierter Inhibitoren, was darauf hindeutet, dass das maschinelle Lernmodell tatsächlich Moleküle entdeckt hatte, die BCL‑2 außer Gefecht setzen könnten. 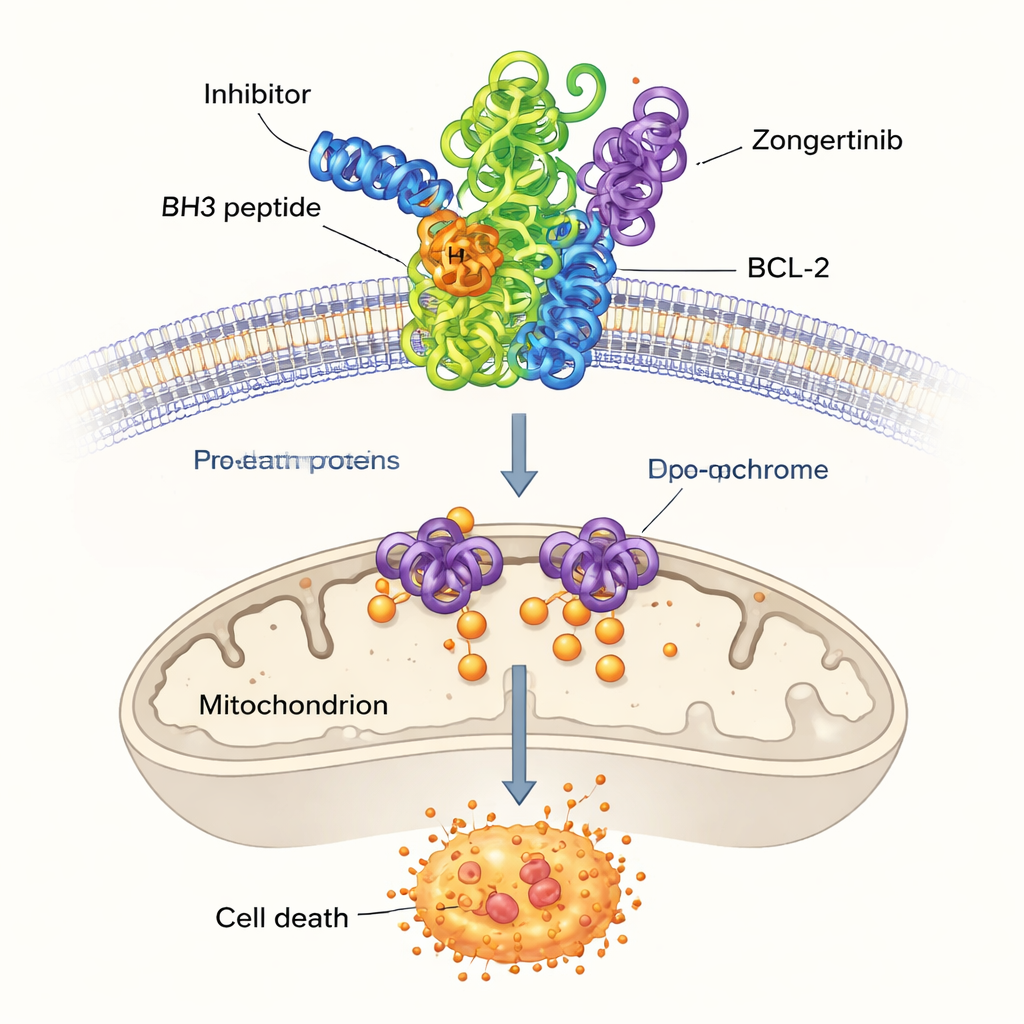
Vorhersagen im Labor prüfen
Computergestützte Hinweise sind nur nützlich, wenn sie unter realen Bedingungen standhalten. Das Team testete daher die drei Kandidaten in einem biochemischen Test, der misst, wie gut eine Verbindung verhindern kann, dass BCL‑2 an einen seiner natürlichen Partner bindet. In einem Konzentrationsbereich zeigte Dersalazine kaum Wirkung. Opelconazole und Zongertinib reduzierten hingegen bei hohen Dosen die BCL‑2‑Aktivität, wobei Opelconazole das Signal nahezu zum Erliegen brachte. Zwar liegen diese Konzentrationen über dem, was für ein kliniktaugliches Medikament ideal wäre, doch sie zeigen, dass die Kandidaten tatsächlich mit BCL‑2 interagieren und validieren die gesamte Entdeckungs‑Pipeline.
Was das für zukünftige Krebstherapien bedeutet
Für Nicht‑Fachleute ist die Kernbotschaft, dass die Forschenden erfolgreich ein Computersystem trainiert haben, zu erkennen, wie ein BCL‑2‑blockierendes Molekül „aussieht“, und es dann nutzten, um eine große Bibliothek vorhandener Medikamente und medikamentenähnlicher Verbindungen zu durchsuchen. Der Ansatz entdeckte bekannte BCL‑2‑Wirkstoffe erneut und hob neue Kandidaten hervor, von denen zwei in Labortests reale Hemmaktivität zeigten. Obwohl noch viel Arbeit nötig ist — Verbesserung der Potenz, Klärung der Sicherheit und Tests in Zellen und Tieren — zeigt diese Studie, wie maschinelles Lernen und kluge Datenaufbereitung die Suche nach besseren Krebsmedikamenten beschleunigen können, indem bereits bekannte Verbindungen wiederverwendet und neu bewertet werden.
Zitation: Park, J., Cho, S., Lee, H. et al. DrugBank mining with machine learning reveals novel candidates for BCL-2 inhibition. Sci Rep 16, 5482 (2026). https://doi.org/10.1038/s41598-026-35117-0
Schlüsselwörter: BCL‑2‑Inhibitoren, maschinelles Lernen, Repositionierung von Arzneimitteln, Apoptose, Krebstherapie