Clear Sky Science · de
Ein physik-informiertes Graph‑Neural‑Network zur Annäherung docking-basierter Bindungsaffinitäten für DYRK2 in der Alzheimer‑Medikamentenrepositionierung
Warum das für Alzheimer wichtig ist
Die Alzheimer‑Krankheit nimmt weltweit zu, doch die meisten verfügbaren Medikamente lindern nur Symptome, anstatt die Erkrankung aufzuhalten. Die Laborprüfung neuer Wirkstoffe ist zeitaufwendig und teuer, besonders für weniger erforschte Gehirnproteine, die für Gedächtnis und neuronale Gesundheit wichtig sein könnten. Diese Studie untersucht eine kluge Abkürzung: ein physikbewusstes KI‑Modell zu verwenden, um vorherzusagen, wie gut bereits zugelassene Alzheimer‑Medikamente an ein wenig untersuchtes Protein namens DYRK2 binden könnten — ein Weg, der neue Behandlungsmöglichkeiten eröffnen könnte.
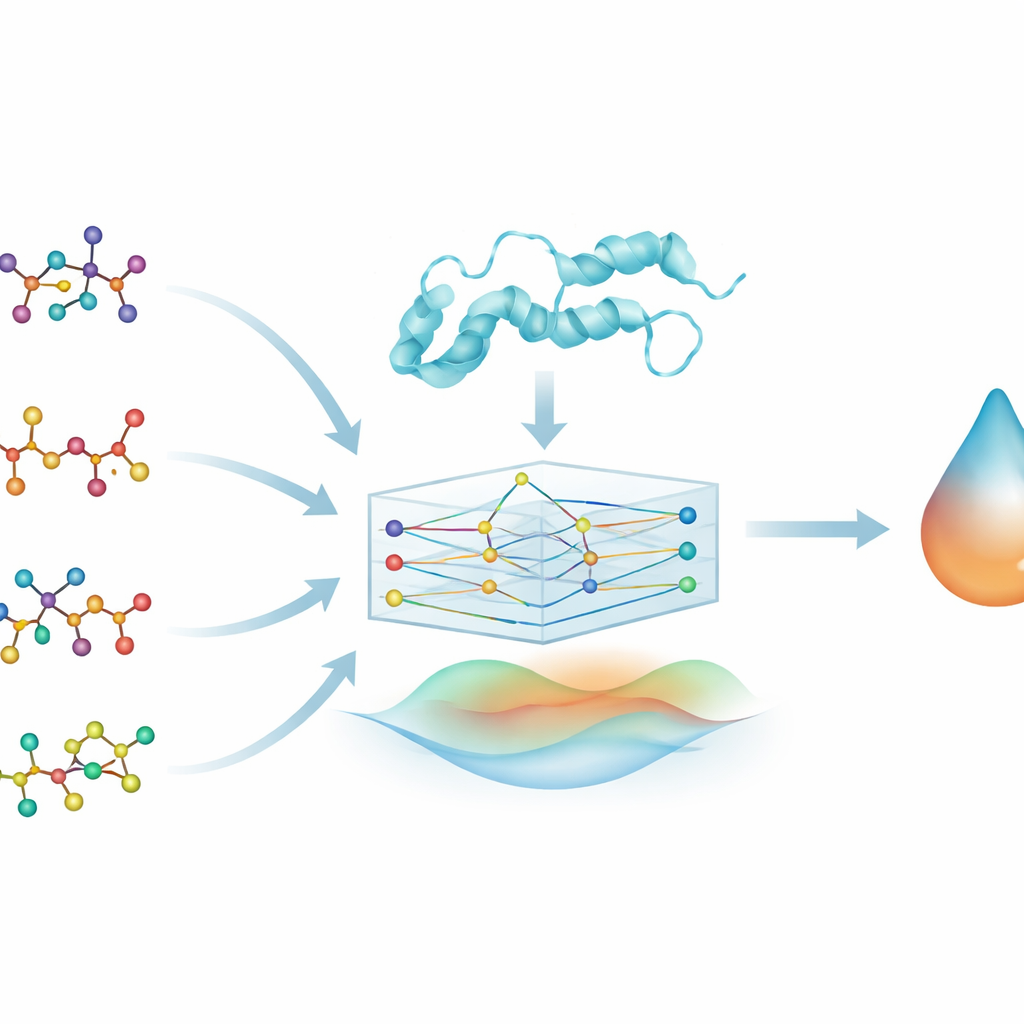
Eine neue Sicht auf alte Medikamente
Statt völlig neue Verbindungen von Grund auf zu entwerfen, konzentrieren sich die Forschenden auf Medikamentenrepositionierung — also neue Anwendungen für bereits zugelassene und weitgehend als sicher bekannte Arzneimittel. Sie untersuchen vier vertraute Alzheimer‑Medikamente (Brexpiprazol, Donepezil, Galantamin und Rivastigmin) und fragen, wie stark jedes an DYRK2 bindet, einer Proteinkinase, die an Wachstum und Funktion von Nervenzellen beteiligt ist. DYRK2 wurde in der Alzheimer‑Forschung kaum betrachtet, aber erste Hinweise verbinden es mit Synapsen, Axonen und Gedächtnis, was es zu einem interessanten Ziel macht, das bestehende Therapien ergänzen könnte.
Moleküle als Netzwerke darstellen
Um die Beziehungen zwischen Medikamenten und Protein zu erforschen, wandelt das Team jedes Wirkstoffmolekül in einen Graphen um: Atome werden zu Punkten und chemische Bindungen zu verbindenden Linien. Ähnlich wird DYRK2 als Kette verbundener Einheiten aus Aminosäuren dargestellt. Ein Maschinlern‑Modelltyp, das Graph‑Neural‑Network (GNN), kann natürlich mit solchen graphförmigen Eingaben arbeiten, Informationen entlang der Verbindungen weiterleiten und Muster in Form und Chemie lernen. So kann das Modell, genannt PhysDual‑GCN, sowohl das Medikament als auch DYRK2 als interagierende Netzwerke „lesen“ statt als einfache Zeichenketten oder Merkmalslisten.
Physik mit künstlicher Intelligenz verbinden
Die meisten Deep‑Learning‑Werkzeuge in der Wirkstoffforschung lernen nur aus Daten, wodurch ihre inneren Mechanismen schwer zu interpretieren sind. Hier verweben die Autorinnen und Autoren bewusst grundlegende physikalische Konzepte darüber, wie Atome wechselwirken. Neben den gelernten Graph‑Merkmalen berechnet PhysDual‑GCN zwei klassische Energiebegriffe: einen, der elektrische Anziehung und Abstoßung zwischen partiellen Ladungen erfasst, und einen weiteren, der die Wechselwirkung durch van‑der‑Waals‑Kräfte beschreibt. Diese physikbasierten Energien werden mit der internen Repräsentation des GNN kombiniert, bevor es eine vorhergesagte Bindungsstärke ausgibt. Effektiv wird das Modell darauf trainiert, das Verhalten klassischer Docking‑Programme — insbesondere AutoDock Vina und verwandte Werkzeuge — nachzuahmen, jedoch schneller und verankert in vertrauten physikalischen Prinzipien.
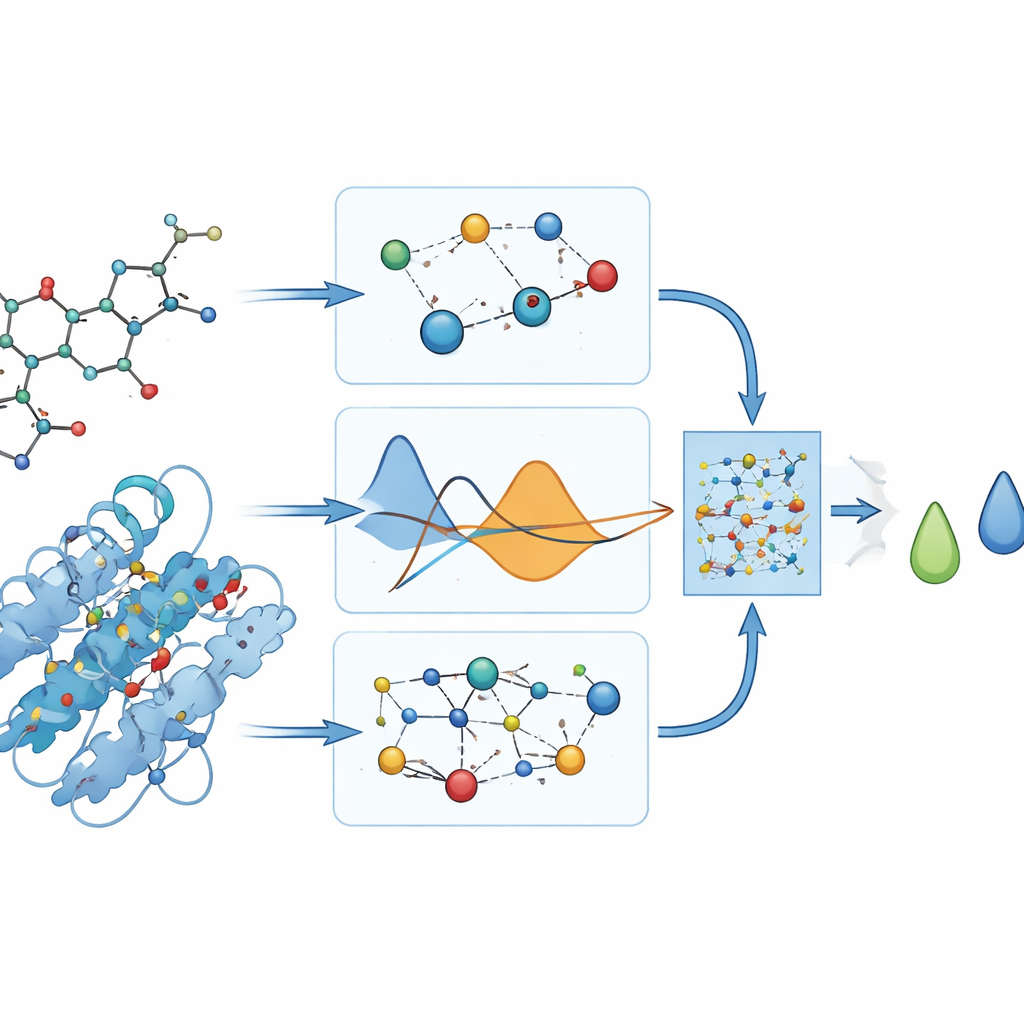
Was das Modell tatsächlich vorhersagt
Da es keine Laborwerte dafür gibt, wie stark diese Medikamente an DYRK2 binden, stützen sich die Autorinnen und Autoren auf Docking‑Programme, die „Referenz“‑Bindungswerte in Energieeinheiten liefern. Sie vermeiden es sorgfältig, diese Werte in den Trainingsprozess einzuspeisen, und nutzen sie stattdessen nachträglich, um zu beurteilen, wie gut PhysDual‑GCN gelernt hat. Für die vier Alzheimer‑Medikamente reproduziert das Modell die Docking‑Werte mit geringen mittleren Fehlern (etwa ein Drittel Kilokalorie pro Mol) und ordnet die Verbindungen korrekt: Donepezil und Brexpiprazol erscheinen als stärkste Binder, während Galantamin und Rivastigmin schwächer, aber noch relativ stabil wirken. Diese Ergebnisse zeigen, dass das physik‑informierte GNN als rechnerischer Ersatz für langsamere Docking‑Berechnungen dienen kann.
Versprechen und Grenzen des Ansatzes
Trotz dieser ermutigenden Zahlen betonen die Autorinnen und Autoren die engen Grenzen ihrer Studie. Es wurden nur vier Medikamente untersucht, und alle Auswertungen beruhen auf anderen Computerprogrammen statt auf realen biochemischen Experimenten. Das DYRK2‑Protein wird vorwiegend als eindimensionaler Sequenzgraph modelliert, nicht als vollständige dreidimensionale Struktur, sodass das Modell noch nicht die detaillierte Form von Bindungstaschen berücksichtigen kann. Auch die physikalischen Energien sind vereinfacht, mit standardisierten Kraftfeldparametern und Schranken. Daher ist die Arbeit als Proof‑of‑Concept zu verstehen: Sie zeigt, dass physikgeführte Graph‑Neural‑Networks klassische Docking‑Werte in datenarmen Szenarien eng nachbilden können, aber nicht, dass die Vorhersagen bereits im Reagenzglas oder in klinischen Studien mit der Realität übereinstimmen.
Was das für die künftige Alzheimer‑Forschung bedeutet
Für Nicht‑Spezialistinnen und Nicht‑Spezialisten lautet die Kernbotschaft, dass intelligente, physikbewusste Algorithmen Forschenden helfen können, neue Alzheimer‑Ziele wie DYRK2 deutlich schneller zu erkunden als traditionelle Methoden allein. Indem PhysDual‑GCN Donepezil und Brexpiprazol als vielversprechende DYRK2‑Binder hervorhebt und einen transparenten Weg zur Annäherung an Docking‑Ergebnisse bietet, liefert es einen Ausgangspunkt für tiefere Laboruntersuchungen. Mit größeren Wirkstoffbibliotheken, reichhaltigeren 3D‑Proteininformationen und experimenteller Validierung könnte ein derartiges Modell zu einem praktischen Werkzeug für das Screening von Kandidatenmedikamenten und die Lenkung von Repositionierungsbemühungen werden, die darauf abzielen, den Verlauf der Alzheimer‑Krankheit zu verlangsamen oder zu verändern.
Zitation: Gider, V., Budak, C. A physics-informed graph neural network to approximate docking-based binding affinity for DYRK2 in Alzheimer’s drug repurposing. Sci Rep 16, 8357 (2026). https://doi.org/10.1038/s41598-026-35102-7
Schlüsselwörter: Alzheimer‑Krankheit, Medikamentenrepositionierung, Graph‑Neural‑Networks, Protein‑Ligand‑Bindung, DYRK2‑Kinase