Clear Sky Science · de
Metagenomisches Sequenzieren identifiziert potenzielle Atemwegserreger in PCR-negativem Teil der Überwachungsproben
Warum versteckte Erreger für alle wichtig sind
Wenn Sie Halsschmerzen oder Husten bekommen, verlassen sich Ärztinnen und Ärzte oft auf schnelle Labortests, um die üblichen Verdächtigen wie Influenza oder COVID-19 nachzuweisen. Was aber passiert, wenn diese Tests „nichts gefunden“ melden, obwohl Sie sich eindeutig krank fühlen? Diese Studie blickt hinter diesen Vorhang, indem sie einen leistungsfähigen DNA-/RNA-basierten Ansatz verwendet, um nach Erregern zu suchen, die Standardtests übersehen, und so ein komplexeres Bild von Atemwegsinfektionen und ihrer Überwachung in der Zukunft zeichnet. 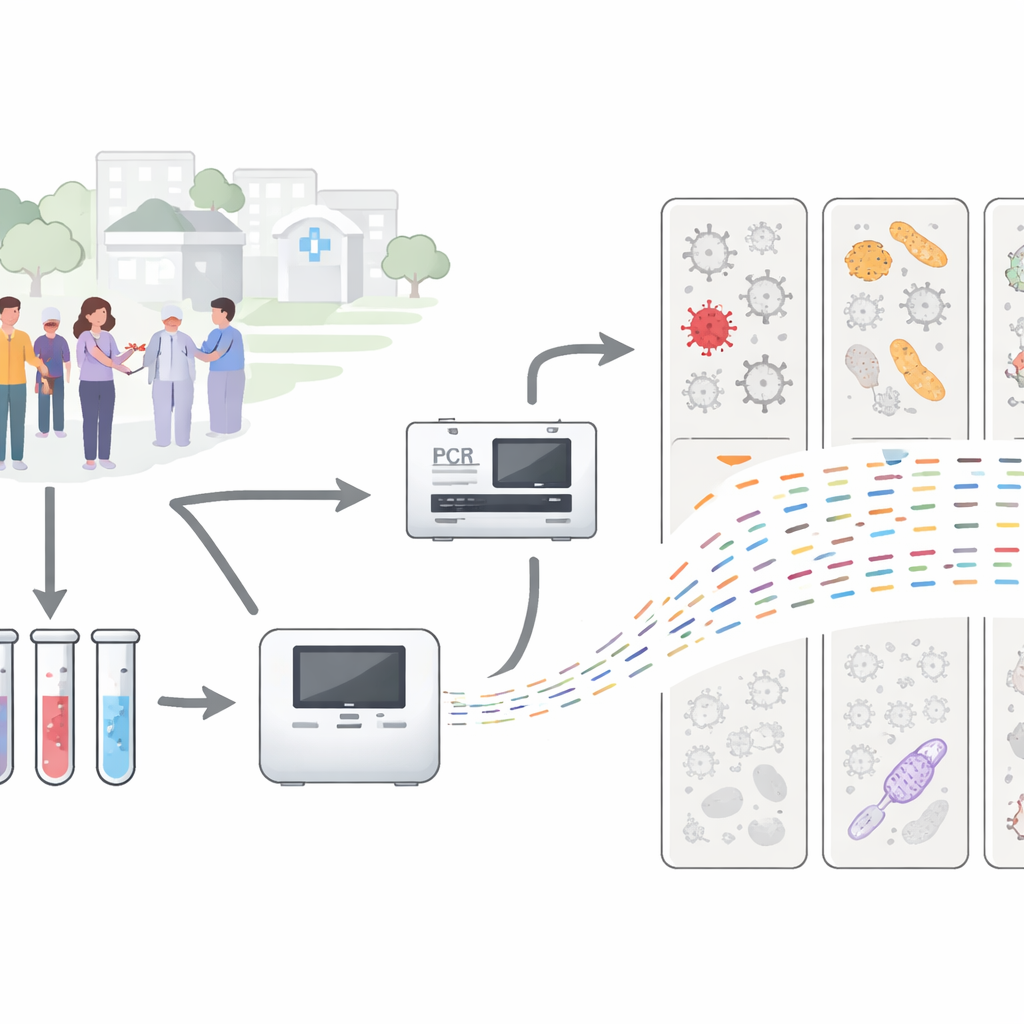
Weiterschauen über das übliche Testpanel hinaus
Während der COVID-19-Pandemie führte Kalifornien ein großes Programm zur Überwachung von Atemwegsinfektionen bei Patientinnen und Patienten in Kliniken mehrerer Landkreise durch. Jede Nasen- oder Rachenprobe wurde mit gängigen Labor-Panels auf eine feste Liste von Viren und Bakterien getestet, zusätzlich zu einem separaten Test auf SARS-CoV-2. Mehr als die Hälfte dieser Proben fiel für jeden Erreger auf der Liste negativ aus, obwohl die Patientinnen und Patienten eindeutige Erkältungs- oder grippeähnliche Symptome hatten. Die Forschenden hinter dieser Arbeit untersuchten 305 dieser „rätselhaften“ Proben sowie 26 bereits als positiv bekannte Proben genauer, um zu sehen, ob fortgeschrittenes Sequenzieren offenlegt, was wirklich vorhanden war.
Alles genetische Material in einer Probe lesen
Anstatt zu fragen „Ist Virus X vorhanden?“, verwendete das Team metagenomisches Sequenzieren, das im Grunde fragt: „Welches genetische Material ist in dieser Probe, ganz gleich, was es ist?“ Sie extrahierten zunächst gesamte DNA und RNA aus jedem Abstrich, vervielfältigten das Material, damit genug für die Analyse vorlag, und führten es dann in Hochdurchsatz-Sequenziermaschinen ein. In einem Teil der Proben fügte man einen zusätzlichen Schritt mit einem „Probe-Capture“-Panel hinzu, das darauf ausgelegt ist, virales genetisches Material herauszufischen, wodurch Viren leichter zu erkennen sind, die sonst durch reichlich menschliches oder bakteriales Material überdeckt würden. Anschließend verglichen Computerprogramme Millionen kurzer genetischer Schnipsel mit großen Referenzdatenbanken, um zu bestimmen, welche Viren, Bakterien und Pilze vorhanden waren. 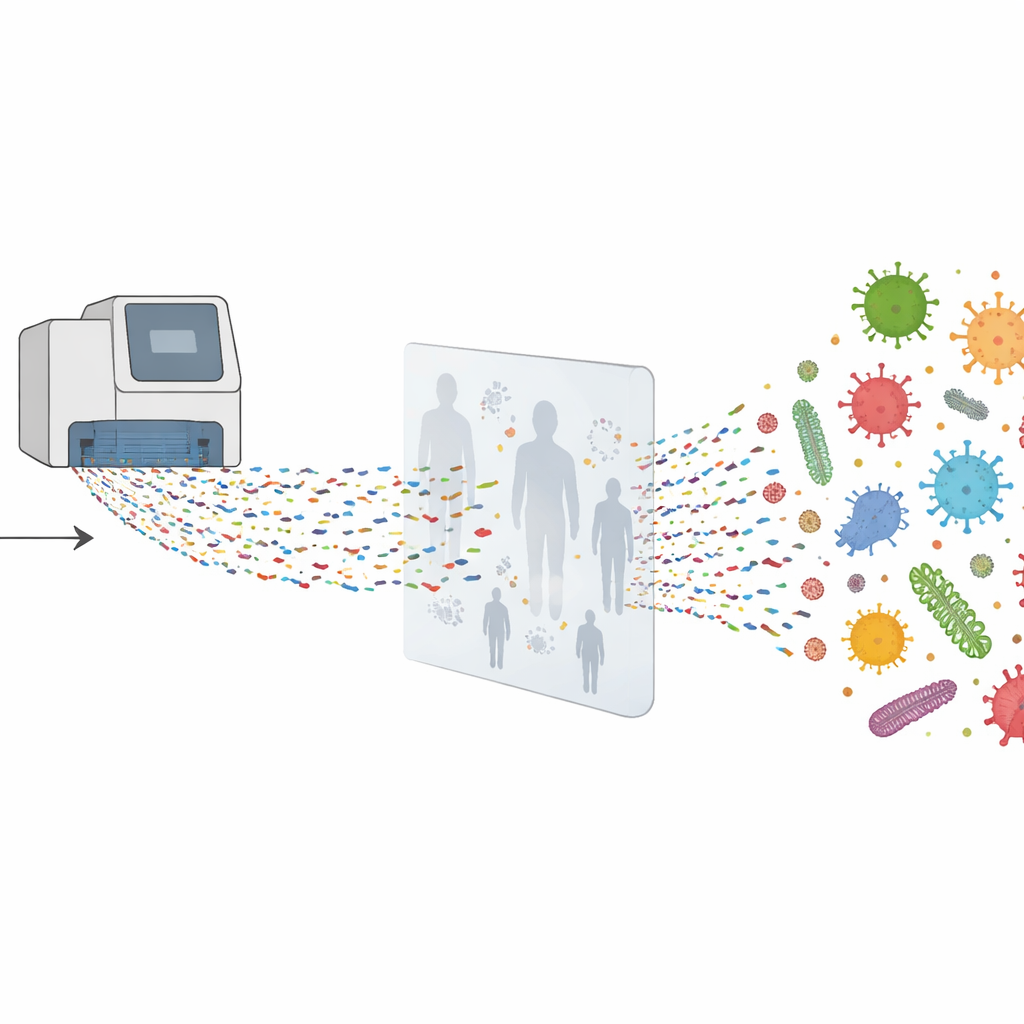
Übersehene Viren und Mikroben aufdecken
Selbst unter Proben, die mit Routinemethoden negativ getestet worden waren, fand der Sequenzieransatz bei etwa fünf Prozent der Fälle humane Atemwegsviren. Dazu gehörten Influenza-C-Virus, humane Bocaviren, Rhinoviren und sogar einige SARS-CoV-2-Infektionen, die Standardtests übersehen hatten. Für viele dieser Viren rekonstruierte das Team nahezu vollständige Genome, wodurch sich zeigen ließ, wie eng verwandt die Stämme untereinander und mit Viren aus anderen Regionen und Jahren waren. Sie fanden auch Proben, die von einer einzigen Bakterien- oder Pilzart dominiert waren, etwa bestimmte Moraxella-, Pseudomonas- oder Penicillium-Arten, was auf eine mögliche bakterielle oder mykotische Beteiligung an Atemwegserkrankungen hindeutet oder zumindest darauf, dass diese Mikroben die lokale mikrobiellen Gemeinschaft in den Atemwegen beeinflussen könnten.
Was verpasste Infektionen uns lehren können
Durch die Rekonstruktion ganzer Virengenome konnten die Forschenden zum Beispiel feststellen, dass die Bocavirus-Stämme in benachbarten Landkreisen nahezu identisch waren, was auf lokale Ausbreitung hindeutet, und dass jede Rhinovirus-Infektion in der Regel einen eigenen, unterschiedlichen Stamm beinhaltete, darunter einen, der eng verwandt mit einem kürzlich beschriebenen neuen Typ war. Sie sahen auch, wie der Virusanreicherungsschritt Menge und Vollständigkeit des viralen genetischen Materials erhöhte, insbesondere bei schwerer zu detektierenden Viren wie Influenza C. Gleichzeitig zeigten viele negative Proben weiterhin keinen eindeutigen Erreger, was betont, dass einige Atemwegssymptome von nicht-infektiösen Ursachen, minderwertigen Proben oder von Mikroben in so geringen Mengen herrühren können, dass sie nicht nachgewiesen werden.
Was das für die künftige Gesundheitsüberwachung bedeutet
Für die routinemäßige klinische Versorgung werden schnelle, zielgerichtete Tests vermutlich weiterhin die Arbeitspferde bleiben: Sie sind günstiger, schneller und leichter durchführbar als Sequenzierung. Diese Studie zeigt jedoch, dass, wenn diese Tests leer ausgehen — besonders bei schweren oder ungeklärten Fällen — ein breit angelegtes metagenomisches Sequenzieren versteckte Infektionen aufdecken, seltene oder ungewöhnliche Viren identifizieren und vollständige Genome für die Verfolgung von Varianten im Zeitverlauf liefern kann. Wenn die Technologie erschwinglicher und standardisierter wird, könnte sie eine kraftvolle Ergänzung zu Routinetests werden und Gesundheitsbehörden helfen, neue Bedrohungen früh zu erkennen und besser zu verstehen, wie eine Vielzahl von Viren, Bakterien und Pilzen in unseren Gemeinschaften zirkuliert.
Zitation: Mascarenhas, A.C., Kantor, R.S., Thissen, J. et al. Metagenomic sequencing identifies potential respiratory pathogens in PCR-negative subset of surveillance samples. Sci Rep 16, 9308 (2026). https://doi.org/10.1038/s41598-025-33917-4
Schlüsselwörter: Atemwegsinfektionen, metagenomisches Sequenzieren, Virusüberwachung, diagnostische Tests, Erregerentdeckung