Clear Sky Science · de
Annotation von 200 Insektengenomen mit BRAKER für konsistente Vergleiche zwischen Arten
Warum Insekten‑Genome wichtig sind
Insekten prägen unsere Welt: Sie bestäuben Nutzpflanzen, übertragen Krankheiten, recyceln Nährstoffe und inspirieren neue Materialien und Technologien. Heute können wir die DNA von tausenden Insektenarten lesen, aber bloß die Genome zu haben reicht nicht aus. Wir brauchen auch eine eindeutige Karte, wo jedes Gen liegt und welche Funktion es wahrscheinlich hat. Dieser Artikel beschreibt ein großes, standardisiertes Projekt zur Annotation der Gene von 200 Insektenarten mit einem automatisierten Ablauf namens VARUS‑BRAKER, wodurch es für Wissenschaftler deutlich einfacher wird, Arten zu vergleichen und nachzuvollziehen, wie Insekten ihre bemerkenswerte Diversität entwickelt haben.
Das Problem unvollständiger genetischer Karten
In den letzten zwei Jahrzehnten ist die Sequenzierung von Insektengenomen von etwa zwanzig Arten auf mehr als viertausend angewachsen. Dennoch besitzt nur etwa eines von zehn Genomen in öffentlichen Datenbanken eine ordentliche Genannotation. Selbst wenn Annotationen vorhanden sind, wurden viele vor Jahren mit älteren Methoden und begrenzten Daten erstellt. Verschiedene Forschergruppen nutzten oft unterschiedliche Software und Evidenzquellen, was künstliche Unterschiede erzeugen kann: Ein Gen kann in einer Art fehlend oder ungewöhnlich geformt erscheinen, nur weil es mit einem anderen Werkzeug annotiert wurde. Dieses Flickwerk an Methoden macht es riskant, Rückschlüsse darauf zu ziehen, wie sich Insektengene wirklich zwischen Arten unterscheiden.
Ein Ein‑Knopf‑Workflow für viele Arten
Die Autorinnen und Autoren begegnen diesem Engpass, indem sie einen automatisierten Ablauf aufbauen, der sich um die Genvorhersage‑Pipeline BRAKER3 zentriert. Ihr VARUS‑BRAKER‑System ist so konzipiert, dass im einfachsten Modus ein Nutzer nur den wissenschaftlichen Namen einer Art angeben muss. Der Workflow lädt dann automatisch das beste verfügbare Genom aus öffentlichen Archiven herunter, sammelt passende RNA‑Sequenzdaten, die zeigen, welche Gene aktiv sind, und ruft Proteininformationen verwandter Arten ab. Er maskiert repetitive DNA, aligniert RNA‑Reads am Genom und kombiniert RNA‑ und Protein‑„Hinweise“, um seine Modelle zu lehren, wo Gene wahrscheinlich beginnen, enden und gespleißt werden. Qualitätsprüfungen wie BUSCO und OMArk bewerten anschließend, wie vollständig und sauber der resultierende Genensatz ist.
Eine weite Reise durch den Insektenbaum
Mithilfe dieses Systems annotierte das Team 200 Insektengenome, die so ausgewählt wurden, dass sie die Hauptzweige des Insektenstammbaums abdecken, mit Fokus auf holometabole Insekten — solche mit vollständiger Metamorphose von Larve über Puppe zum Erwachsenen — sowie einer vielfältigen Auswahl von Verwandten. Ihre Stichprobe umfasst 77 Familien und 14 Ordnungen, darunter Fliegen, Schmetterlinge, Käfer, Bienen, Ameisen, Blattläuse, Kakerlaken und andere. Bei 85 dieser Arten gab es zuvor keine Annotation in GenBank. Für jede Art sagte der Workflow proteinkodierende Gene voraus und lieferte über 4,2 Millionen Proteinsequenzen. Die meisten Genome und ihre vorhergesagten Proteome bestanden strenge Vollständigkeitstests und erreichten typischerweise mindestens 85–95 % Abdeckung der erwarteten Kern‑Gene, was darauf hinweist, dass der automatisierte Ansatz hochwertige Ergebnisse liefert.
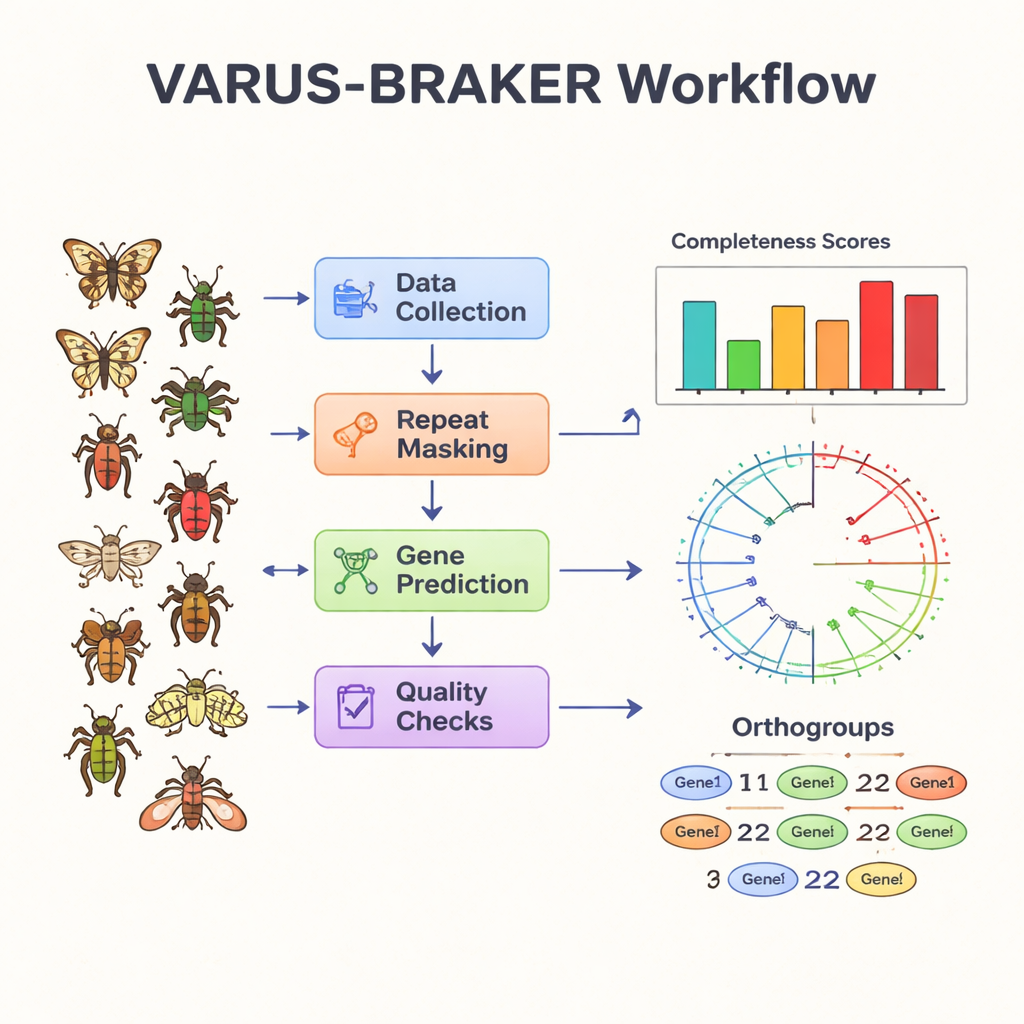
Von Genlisten zur biologischen Bedeutung
Gene aufzulisten ist nur ein Teil der Aufgabe; Forschende benötigen auch Hinweise darauf, was diese Gene tun. Zu diesem Zweck setzten die Autorinnen und Autoren eine funktionelle Annotation‑Pipeline namens FANTASIA ein, die moderne Protein‑Sprachmodelle nutzt, um jedem Protein Gene‑Ontology‑(GO)‑Termini zuzuweisen — standardisierte Bezeichnungen für biologische Funktionen. Im Vergleich zum weit verbreiteten InterProScan‑Tool annotierte FANTASIA etwa 1,6‑mal mehr Proteine, stimmte jedoch eng überein, wenn beide Methoden Vorhersagen machten. Das Team gruppierte außerdem verwandte Gene in „Orthogruppen“, Mengen von Genen mit gemeinsamer Abstammung, und nutzte diese, um einen Evolutionsbaum der 200 Arten zu erstellen. Dieses Framework ermöglicht es zu untersuchen, welche Gene geteilt, verloren oder erweitert wurden in verschiedenen Insektenlinien und Genbestände mit Merkmalen wie Metamorphose oder Larvenverhalten zu verknüpfen.
Eine wiederverwendbare Ressource für zukünftige Entdeckungen
Alle Daten dieses Projekts — einschließlich Genstrukturen, Proteinsequenzen, funktioneller Labels, Orthogruppen, Artbäume und tRNA‑Vorhersagen — sind frei über öffentliche Repositorien verfügbar. Die Autorinnen und Autoren veröffentlichen außerdem den kompletten VARUS‑BRAKER‑Workflow als Open‑Source‑Code, sodass andere Wissenschaftlerinnen und Wissenschaftler neue Insektengenome oder sogar Genome anderer Tiere und Pflanzen konsistent annotieren können. Für Nicht‑Spezialisten lautet die zentrale Aussage: Diese Arbeit verwandelt eine zerstreute Sammlung von DNA‑Sequenzen in einen kohärenten, vergleichbaren Atlas der Insektengene. Mit diesen standardisierten Karten können künftige Studien verlässlicher herausfinden, wie Insekten Flug, Metamorphose und ökologischen Erfolg entwickelt haben, und Gene gezielter für Landwirtschaft, Naturschutz und Krankheitsbekämpfung adressieren.
Zitation: Saenko, S., Hoff, K.J. & Stanke, M. Annotation of 200 Insect Genomes with BRAKER for Consistent Comparisons across Species. Sci Data 13, 288 (2026). https://doi.org/10.1038/s41597-026-06840-0
Schlüsselwörter: Insektengenomik, Genomannotation, vergleichende Genomik, evolutionsbiologie, Bioinformatik