Clear Sky Science · de
Minimales virtuelles Datenset für reproduzierbare de-novo-Assemblierung triploider Genome
Warum dreifach vorhandene Genome wichtig sind
Viele Nutzpflanzen und andere Organismen tragen nicht nur zwei Kopien jedes Chromosoms, wie es beim Menschen der Fall ist, sondern drei oder mehr. Aus den DNA-Sequenzdaten diese zusätzlichen Kopien zusammenzusetzen ist überraschend schwierig, weil die Kopien einander sehr ähnlich, aber nicht identisch sind. Dieser Artikel stellt ein kleines, sorgfältig gestaltetes „virtuelles“ Datenset vor, mit dem Forschende Genom‑Assemblierungsprogramme an einem realistischen dreifach-Kopien‑(triploiden) Problem testen und vergleichen können, unter Bedingungen, die vollständig bekannt und reproduzierbar sind.
Aufbau eines einfachen Stellvertreter‑Genoms
Statt bei einer realen Pflanze oder einem Tier zu beginnen, erzeugt die Autorin zunächst eine zufällige DNA-Sequenz von einer Million Basen als sauberes Ausgangsmodell. Dieses Modell wird dann in drei separate Versionen dupliziert und steht so für die drei Chromosomensätze eines triploiden Organismus. Um nachzuahmen, wie reale Genome sich über Zeit langsam verändern, fügt die Studie in jeder Kopie schrittweise eine feste Anzahl winziger Änderungen—Einzelbasen‑Substitutionen—ein. Wird dieser Prozess über 100 Schritte wiederholt, entstehen Tripel von Genomen, die von nahezu identisch bis deutlich, aber noch mäßig verschieden reichen. Dieser kontrollierte „Divergenzgradient“ bildet das Rückgrat des Benchmarks.
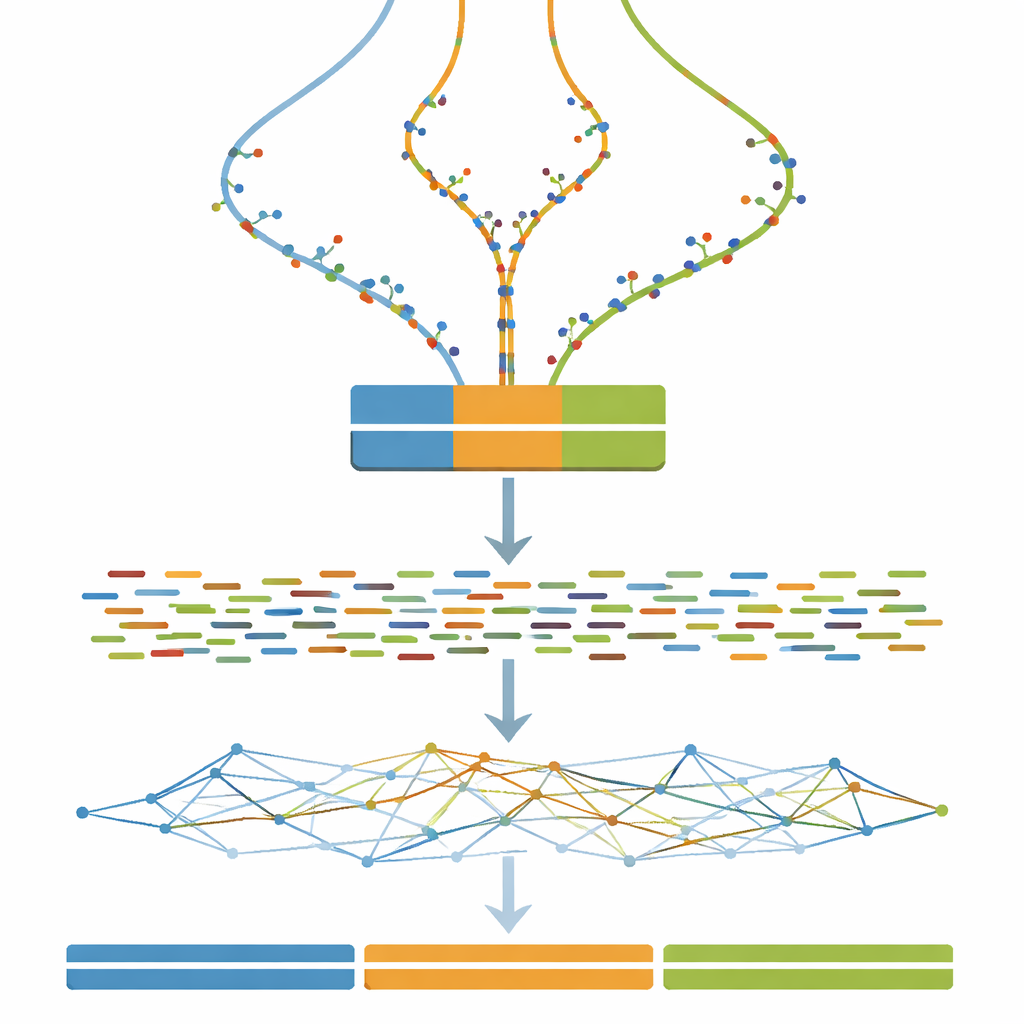
Virtuelle Genome in virtuelle Experimente verwandeln
Sobald jedes dreifache Genom definiert ist, besteht der nächste Schritt darin, zu simulieren, was eine DNA‑Sequenziermaschine sehen würde. Die Studie verwendet weit verbreitete Software, um kurze gepaarte DNA‑Fragmente zu simulieren, ähnlich denen, die ein Illumina‑Sequencer liefert, bei einer konstanten und relativ hohen Abdeckungstiefe. Optionale Aufräumschritte ahmen gängige Praxis nach, etwa das Korrigieren zufälliger Sequenzierfehler und das Zusammenführen überlappender Reads. Dadurch kann man mit dem Datenset nicht nur Assemblierungsalgorithmen testen, sondern auch prüfen, wie typische Vorverarbeitungsentscheidungen die finalen Assemblierungen beeinflussen.
Stresstests für Assemblierungsstrategien
Der Kern der Arbeit ist ein großes Experiment, in dem alle simulierten Reads in ein einziges Genom‑Assemblierungsprogramm eingespeist werden, während nur eine zentrale Einstellung variiert wird: die k‑Mer‑Größe, ein Parameter, der steuert, wie fein die Software die Reads beim Zusammenbau in Stücke „zerlegt“. Für jede Kombination aus Divergenzstufe (von 0 bis 100 Schritten) und k‑Mer‑Größe (ein breites Spektrum ungerader Werte) wird eine neue Assemblierung erstellt. Ein Begleit‑Evaluationswerkzeug misst dann, wie zusammenhängend die zusammengesetzten Stücke sind, wie viele Stücke existieren und wie genau ihre kombinierte Länge der bekannten Wahrheit von drei Millionen Basen entspricht. Diese Messwerte werden als Heatmaps zusammengefasst und zeigen große Bereiche, in denen Assemblierungen verschiedene Kopien zu einer zusammenfallen lassen, in viele kleine Fragmente zerfallen oder dem Ideal nahekommen: drei lange, genaue Contigs.
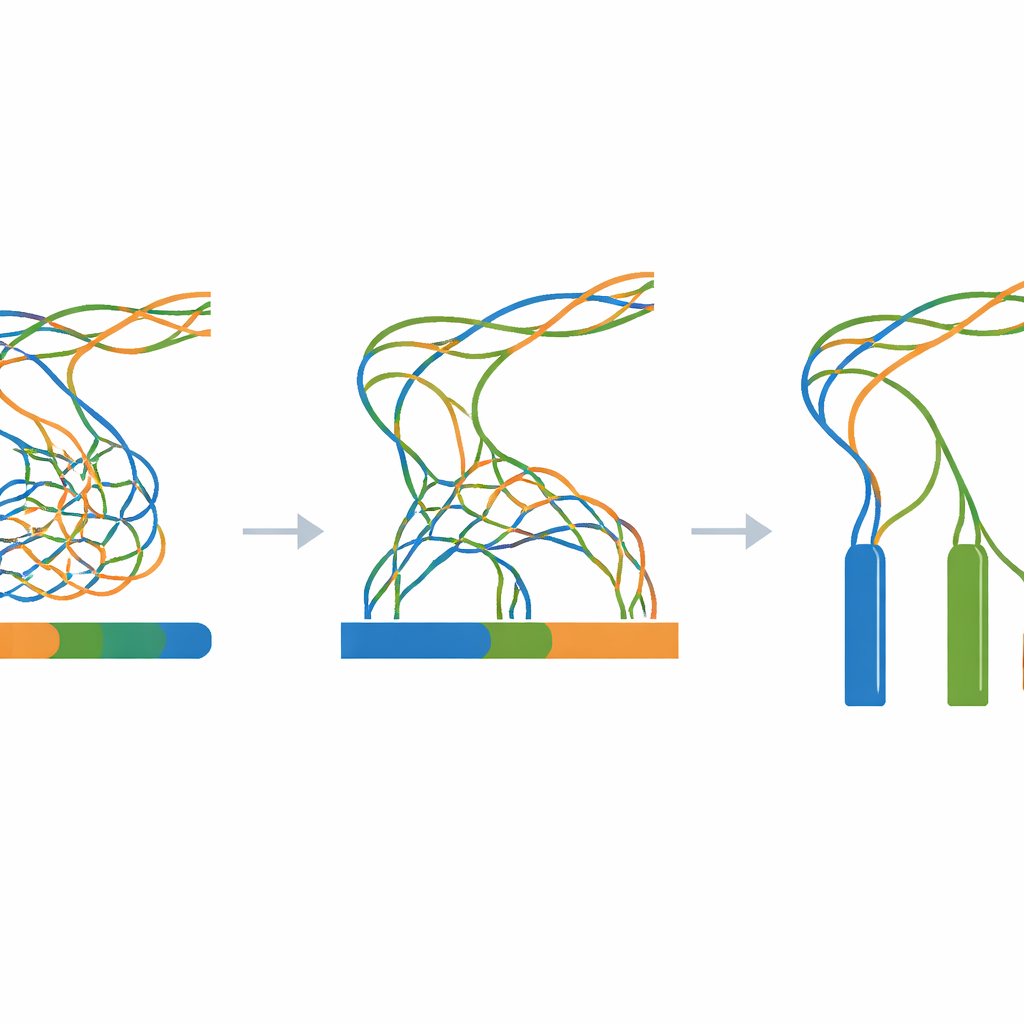
Eine transparente Referenz für schwierige Genome
Da jeder Schritt synthetisch und skriptgesteuert ist—von der anfänglichen zufälligen Vorlage bis zu den finalen Assemblierungen—können Forschende den gesamten Workflow auf jedem Standard‑Linux‑Rechner allein mit Open‑Source‑Werkzeugen reproduzieren. Das in der Arbeit verlinkte Zenodo‑Archiv enthält das Vorlagen‑Genom, alle Zwischen‑Mutationssequenzen, alle simulierten Reads und jede Assemblierungsergebnisdatei sowie Logs und einfache Hilfsskripte. Technische Überprüfungen bestätigen, dass der Mutationsprozess wie erwartet funktioniert, dass die simulierten Reads den geforderten Längen und der Abdeckung entsprechen und dass die Assemblierungen das erwartete Muster zeigen: starkes Überkollabieren, wenn die drei Kopien nahezu identisch sind, und klarere Trennung, wenn sie weiter auseinanderdriften.
Was das in einfachen Worten bedeutet
In einfachen Worten bietet dieser Artikel eine kontrollierte Teststrecke für Software, die versucht, drei ähnliche Anleitungsbücher aus Bergen von durcheinandergeratenen Fragmenten wieder zusammenzusetzen. Indem man schrittweise erhöht, wie unterschiedlich die drei Bücher sind, und systematisch eine zentrale Einstellung im Rekonstruktionsprozess variiert, macht das Datenset leicht sichtbar, wann und wie aktuelle Methoden versagen oder Erfolg haben. Entwicklerinnen und Entwickler können es nutzen, um neue Algorithmen zu optimieren, während Anwenderinnen und Anwender besser verstehen können, welche Einstellungen für triploide Genome am besten funktionieren. Obwohl die DNA selbst künstlich ist, sind die daraus gewonnenen Erkenntnisse—über Kollaps, Trennung und die Auswirkungen von Parameterentscheidungen—direkt relevant für reale Bemühungen, die komplexen Genome vieler wichtiger Arten zu entschlüsseln.
Zitation: Ootsuki, R. Minimum virtual dataset for reproducible triploid de novo genome assembly. Sci Data 13, 382 (2026). https://doi.org/10.1038/s41597-026-06779-2
Schlüsselwörter: triploide Genomassemblierung, Polyploid-Benchmarking, synthetisches DNA-Datenset, de-novo-Assemblierung, k‑Mer-Optimierung