Clear Sky Science · de
Genomsequenzierung, de-novo-Assembly und Annotation des wirtschaftlich wichtigen Bambus Bambusa tulda Roxb
Ein schnell wachsendes Gras mit großem Potenzial
Bambus mag wie eine einfache Gartenpflanze wirken, ist aber in Wirklichkeit eine leistungsfähige natürliche Ressource für Bauholz, Papier und sogar künftige Biokraftstoffe. Eine weit verbreitete Art, Bambusa tulda oder Bengalischer Bambus, wächst schnell, speichert große Mengen an holzigem Material und blüht nur selten. Bislang fehlte Wissenschaftlern ein vollständiges „Bedienhandbuch“ für diese Art. Dieser Artikel beschreibt, wie Forschende die gesamte DNA-Sequenz von B. tulda entschlüsselt und organisiert haben und so eine grundlegende Ressource geschaffen haben, die hilft, Bambus für Industrie, Naturschutz und klimafreundliche Technologien zu verbessern.
Warum die DNA eines Bambus entschlüsseln?
Bambusa tulda ist auf dem indischen Subkontinent und in Teilen Südostasiens verbreitet, wo seine robusten Halme in ländlichen Bauweisen, Möbeln und Handwerk verwendet werden. Er gewinnt auch als Quelle für Zellstoff und erneuerbare Energie an Interesse. Dennoch verhält sich B. tulda rätselhaft: Er kann sehr schnell wachsen, viel zähes Holzmaterial anreichern und dann etwa 50 Jahre warten, bevor er blüht — manchmal tun das alle Pflanzen in einem Gebiet gleichzeitig. Ohne eine vollständige Genomsequenz konnten Forschende nur vermuten, welche Gene diese Merkmale steuern. Durch das Lesen und Zusammenfügen seiner DNA wollten die Autoren eine Referenzkarte erstellen, die künftigen Forschenden erlaubt, Wachstum, Blüte, Krankheitsresistenz und mehr zu untersuchen.
Messung und Lesen eines Riesen-Genoms
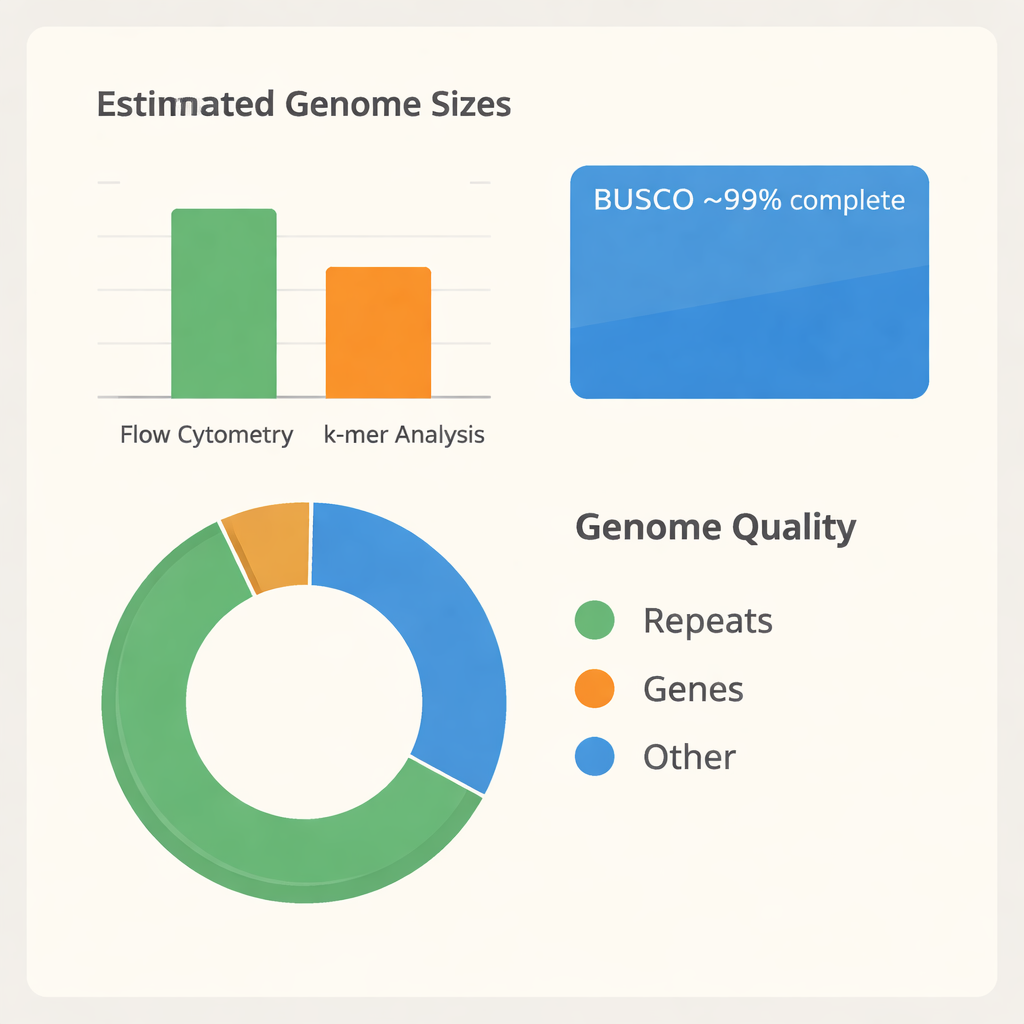
Das Team musste zunächst einschätzen, wie groß das B. tulda-Genom ist. Mithilfe einer Technik namens Durchflusszytometrie verglichen sie den DNA-Gehalt von B. tulda-Blattzellen mit dem von Tomate und Mais, zwei Pflanzen, deren Genomgrößen bereits bekannt sind. Das deutete auf eine diploide Genomgröße von etwa 3 Milliarden DNA-„Buchstaben“ hin. Anschließend nutzten sie einen zweiten, unabhängigen Ansatz, der darauf beruht, wie sich kurze DNA-Fragmentüberlappungen verhalten (k-mer-Analyse). Dieser schätzte eine etwas geringere Größe von etwa 2,34 Milliarden Basen und zeigte, dass ein großer Teil des Genoms repetitiv und wahrscheinlich dupliziert ist. Mit diesen Messwerten extrahierten sie sehr lange, hochwertige DNA aus jungen Blättern und sequenzierten sie mit moderner PacBio-HiFi-Technologie und erzeugten über 116 Milliarden Basen Rohdaten — genug, um das Genom dutzende Male zu lesen.
Das Bambus-Bauplan zusammensetzen
Millionen von DNA-Reads in ein geordnetes Genom zu verwandeln ist wie das Zusammensetzen eines riesigen Puzzles ohne Bildvorlage. Die Forschenden nutzten spezialisierte Software, um sowohl eine kombinierte Primär-Assembly als auch zwei separate Haplotypen zu erstellen, die die beiden elterlichen Genomkopien widerspiegeln. Nach Entfernen doppelter und organell-abgeleiteter Sequenzen erhielten sie eine gestraffte „haploide“ Assembly aus 43 großen Segmenten mit zusammen etwa 1,37 Milliarden Basen. Diese Segmente gliedern sich in drei Subgenome, bezeichnet A, B und C, was mit dem komplexen, polyploiden Ursprung von B. tulda übereinstimmt. Ein weit verbreiteter Qualitätstest namens BUSCO zeigte, dass etwa 99 % der erwarteten Pflanzen-Gene vorhanden und intakt sind, was darauf hindeutet, dass die Assembly sowohl vollständig als auch zuverlässig für weiterführende Studien ist.
Gene, Wiederholungen und evolutionäre Hinweise
Sobald das Genom zusammengebaut war, bestand der nächste Schritt darin, seine funktionalen Teile zu identifizieren. Durch die Kombination dreier Evidenzlinien — Vorhersagen aus der DNA-Sequenz selbst, Ähnlichkeiten zu Genen anderer Bambusse und RNA-Daten aus aktiv exprimierten Genen — annotierte das Team 56.890 proteinkodierende Gene, die etwa ein Fünftel des Genoms einnehmen. Sie katalogisierten außerdem zahlreiche nicht-kodierende RNAs, darunter über tausend Transfer- und ribosomale RNA-Gene, die die Proteinsynthese unterstützen. Auffällig ist, dass etwa zwei Drittel des Genoms aus repetitiven Elementen bestehen, insbesondere aus mobilen DNA-Abschnitten, die sich kopieren und verschieben. Diese Wiederholungen erklären, warum frühere Größenschätzungen variierten, und weisen auf eine dynamische evolutionäre Geschichte hin. Der Vergleich von Proteinfamilien über zwölf weitere Bambusarten sowie Mais und Banane als Verwandte ordnete B. tulda eindeutig den paleotropischen holzigen Bambussen mit einem hexaploiden Hintergrund zu und bestätigte, dass sein Genom aus mehreren Vorfahrenskopien aufgebaut ist.
Eine neue Grundlage für zukünftige Bambusforschung
Für Nicht-Spezialisten ist das wichtigste Ergebnis, dass B. tulda jetzt eine hochwertige Referenzgenomsequenz besitzt — einen indexierten, durchsuchbaren Bauplan seiner DNA. Diese Ressource ermöglicht es Forschenden, Gene zu identifizieren, die schnelles Wachstum, Holzigkeit und verzögerte Blüte steuern, und sie mit denen anderer Süßgräser zu vergleichen. Sie wird auch Bemühungen unterstützen, Bambussorten zu züchten oder zu entwickeln, die besser für Bau, Papier oder Energie geeignet sind, während natürliche Populationen erhalten bleiben. Kurz gesagt: Durch die Kartierung der genetischen Landschaft dieses wirtschaftlich wichtigen Bambus legt die Studie das Fundament für eine klügere Nutzung einer der vielseitigsten Pflanzen der Welt.
Zitation: Kundu, S., Rupp, O., Dey, S. et al. Genome sequencing, de novo assembly and annotation of the commercially important bamboo, Bambusa tulda Roxb. Sci Data 13, 175 (2026). https://doi.org/10.1038/s41597-026-06679-5
Schlüsselwörter: Bambus-Genom, Bambusa tulda, Pflanzengenetik, holzige Süßgräser, erneuerbare Biomaterialien