Clear Sky Science · de
Eine Chromosomen‑Level-Genomassemblierung von Homatula variegata aus dem Einzugsgebiet des Jangtse
Ein winziger Bachfisch mit großer genetischer Geschichte
In den schnell fließenden, kiesigen Bächen, die den oberen Jangtse speisen, lebt ein kleiner, gestreifter Schlammling namens Homatula variegata. Er wird sowohl als Speisefisch als auch für Aquarien geschätzt, doch über seine DNA-Blueprint war bislang fast nichts bekannt. Diese Studie liefert die erste nahezu vollständige, Chromosom für Chromosom aufgebaute Karte des Genoms der Art und ebnet den Weg für klügeren Schutz, effizientere Zucht und ein tieferes Verständnis dafür, wie Leben sich an kalte, schnell fließende Gebirgsgewässer anpasst.
Warum das Genom eines unscheinbaren Fisches kartieren?
Obwohl dieser Schlammling nur etwa 14 Zentimeter erreicht, spielt er eine überproportionale Rolle in lokalen Flusssystemen und der regionalen Aquakultur. Er gedeiht in mittelhohen Bächen mit kiesigem Grund und stetiger Strömung und ernährt sich von Insekten, organischen Partikeln und kleinen Fischen. Da er sowohl schmackhaft als auch farbenfroh ist, wächst das Interesse, ihn als heimische Zier- und Spezies zu züchten. Zucht und Schutz einer Art sind jedoch deutlich schwieriger ohne eine präzise genetische Referenz. Ein vollständiges Genom wirkt wie eine detaillierte Ersatzteilliste und ein Schaltplan: Es offenbart Gene, die Wachstum, Färbung, Krankheitsresistenz und die Fähigkeit steuern, mit schnellem, kühlem Wasser zurechtzukommen. Bislang hatte keine Art dieses Zweigs der Schlammlinge eine derart hochwertige Referenz, wodurch ein bedeutendes Blindfeld in der Genetik von Süßwasserfischen bestand.
Aufbau einer Chromosomen-für-Chromosomen-DNA-Karte
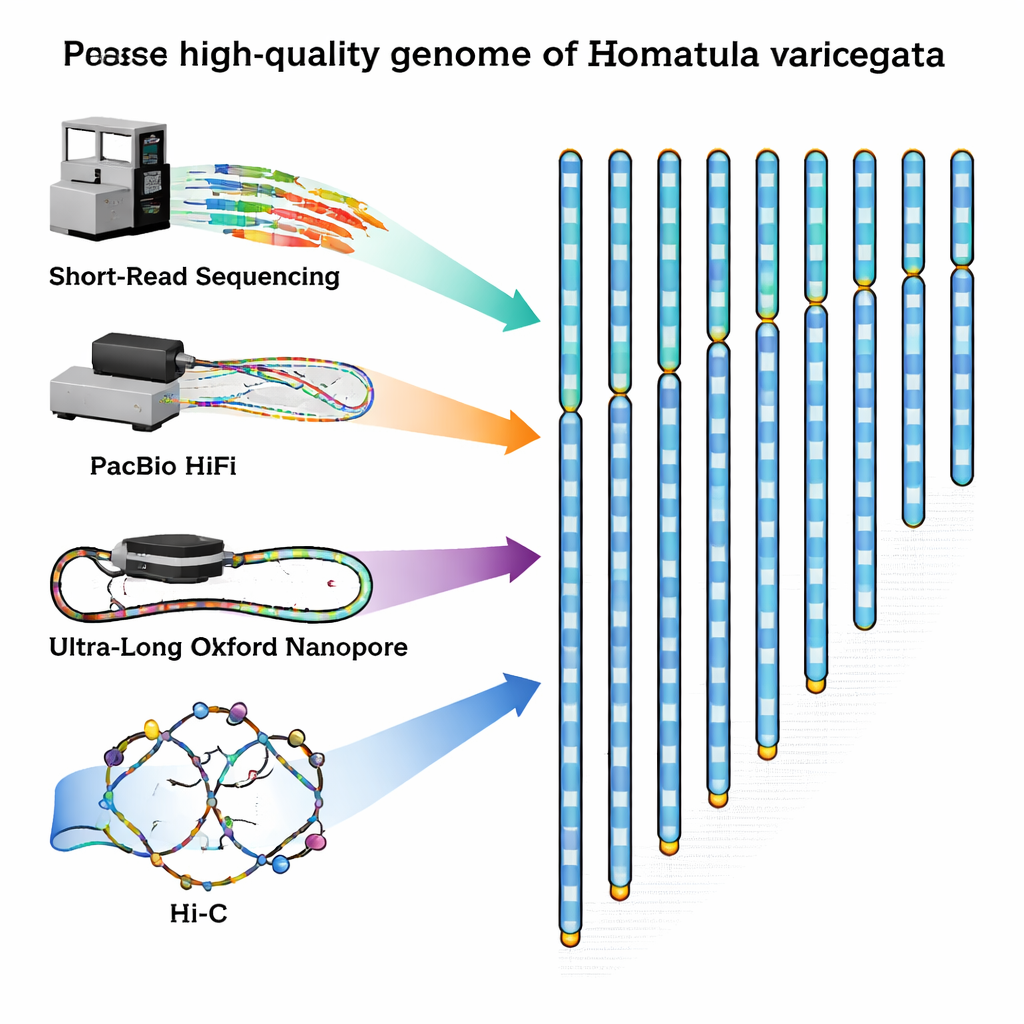
Um diese Lücke zu schließen, fing das Forscherteam einen gesunden erwachsenen Männchen aus dem Qingyi-Fluss, einem Nebenfluss des Jangtse, und isolierte sorgfältig DNA und RNA aus dessen Blut. Anschließend kombinierten sie mehrere moderne Sequenzierverfahren, die jeweils unterschiedliche Stärken haben. Kurzread-Geräte von Illumina erzeugten riesige Mengen hochpräziser DNA-Schnipsel. Die PacBio-HiFi-Technologie lieferte etwas längere Fragmente mit ausgezeichneter Genauigkeit, während Oxford-Nanopore-Geräte ultralange Stränge erzeugten, die über schwer zu entschlüsselnde, repetitive Regionen hinwegreichen können. Schließlich erstellte eine Methode namens Hi‑C eine 3-D-Kontaktkarte, die zeigt, wie DNA-Stränge im Zellkern gefaltet und miteinander in Kontakt stehen, und half so, Fragmente in der richtigen Reihenfolge zu vollständigen Chromosomen zu verbinden.
Was das neue Genom offenbart
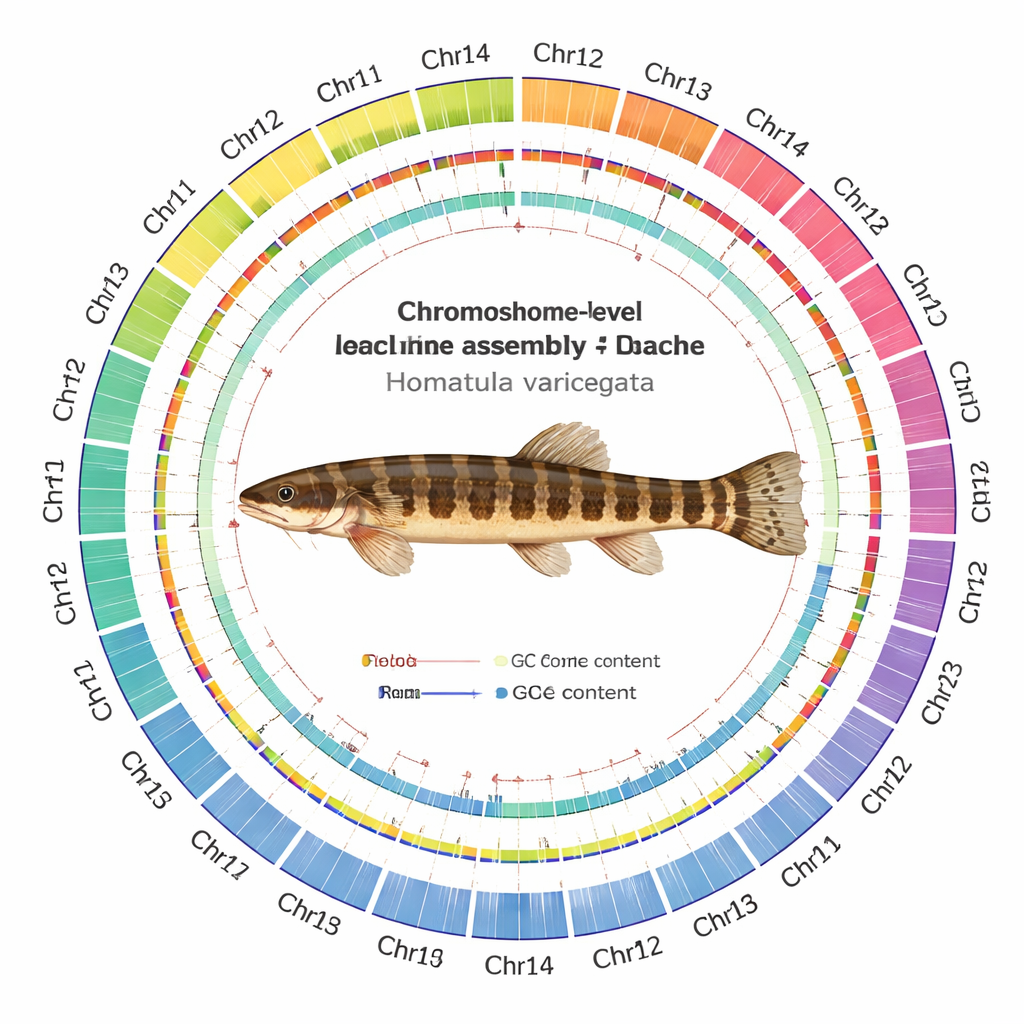
Durch das Verweben dieser Daten mit moderner Assemblierungssoftware und gründlichen Qualitätsprüfungen erzeugte das Team ein Genom mit 641 Millionen Basenpaaren, sauber organisiert in 24 Chromosomen. Bemerkenswerterweise ist jedes Chromosom als ein einzelnes, kontinuierliches Stück assembliert, wobei 22 Chromosomen überhaupt keine Lücken aufweisen und nur zwei sehr kleine Lücken enthalten. Die Forscher konnten 24 wahrscheinliche Zentromere lokalisieren — den zentralen „Gürtel“ jedes Chromosoms — und die meisten der schützenden Telomer‑Kappen an den Chromosomenenden nachweisen. Sie katalogisierten 24.479 proteinkodierende Gene und konnten etwa 93 % davon durch Vergleiche mit großen internationalen Datenbanken mit wahrscheinlichen Funktionen belegen. Zudem kartierten sie die Landschaft wiederholter DNA und fanden, dass mehr als ein Viertel des Genoms aus mobilen genetischen Elementen besteht, insbesondere DNA-Transposons, die im Genom springen können und manchmal evolutionäre Veränderungen vorantreiben.
Qualitätstests unter der Haube
Hohe Gesamtzahlen sind nur aussagekräftig, wenn die zugrunde liegende Karte vertrauenswürdig ist. Daher unterzogen die Forschenden die Assemblierung einer Reihe von Tests. Reads aller Sequenzierplattformen ließen sich mit sehr hoher Rate auf das neue Genom zurückalignieren, mit gleichmäßiger Abdeckung über den Großteil der Chromosomen. Unabhängige Werkzeuge, die kurze DNA-Muster zählen, deuteten darauf hin, dass nahezu der gesamte erwartete Sequenzinhalt vorhanden ist, und Standardtests zur Genvollständigkeit zeigten, dass die überwiegende Mehrzahl universeller Fischgene intakt ist. Die Hi‑C-Kontaktkarten bildeten saubere, quadratische Muster entlang jedes Chromosoms mit wenig Streusignal dazwischen, was darauf hindeutet, dass Segmente korrekt verbunden und nicht durcheinandergebracht wurden.
Von der DNA-Karte zur realen Wirkung
Für einen Nicht‑Spezialisten mag diese Arbeit wie ein technischer Triumph an sich erscheinen, aber ihre Auswirkungen sind praktisch und weitreichend. Ein nahezu durchgehendes Genom von Homatula variegata bietet Forschern eine Referenz, mit der sie Wildpopulationen vergleichen, genetische Vielfalt verfolgen und Anzeichen von Inzucht oder lokaler Anpassung erkennen können. Züchter können nach DNA-Markern suchen, die mit erwünschten Merkmalen wie schnellem Wachstum, Widerstandsfähigkeit oder auffälliger Färbung verknüpft sind, was selektive Züchtung beschleunigt und zugleich den natürlichen Charakter der Art bewahrt. Ökologen können untersuchen, wie sich dieser Schlammling an das Leben in kühlen, schnell fließenden Gebirgsbächen angepasst hat — Erkenntnisse, die auch das Management verwandter Arten informieren könnten. Kurz: Dieses Chromosomen‑Level-Genom verwandelt einen bescheidenen Flussfisch in ein mächtiges Modell zum Verständnis — und Schutz — des reichen Lebens asiatischer Süßwasserökosysteme.
Zitation: Tang, Y., Wu, Q., Wang, Y. et al. A chromosome level genome assembly of Homatula variegata from the Yangtze River basin. Sci Data 13, 303 (2026). https://doi.org/10.1038/s41597-026-06667-9
Schlüsselwörter: Fischgenom, Chromosomenassemblierung, Süßwasser-Biodiversität, Aquakulturgenetik, Jangtse-Schlammling