Clear Sky Science · de
Erste Chromosomenebenen-Genomassemblierung und -annotation einer bedrohten Süßwasser-Stachelrochenart (Fontitrygon garouaensis) aus Afrika
Ein seltener Flussbewohner rückt genomisch ins Rampenlicht
In den trüben Flüssen Westafrikas lebt ein ungewöhnlicher Stachelrochen, der etwas getan hat, was fast alle seine Hai- und Rochenverwandten nicht geschafft haben: er ist dauerhaft ins Süßwasser gezogen. Dieser glatte Süßwasser-Stachelrochen, Fontitrygon garouaensis, ist zudem bedroht und kaum erforscht. Die Studie, auf der dieser Artikel basiert, liefert die erste vollständige, auf Chromosomenebene aufgelöste Karte seines Erbguts und bietet damit ein mächtiges neues Werkzeug, um zu verstehen, wie sich eine so alte marine Linie an ein Leben in Flüssen angepasst hat und wie wir sie besser schützen können.
Warum dieser Stachelrochen wichtig ist
Haie und Rochen gehören zu einem der ältesten Zweige des Wirbeltierstammbaums und trennten sich vor Hunderten Millionen Jahren von den Vorfahren der Knochenfische. Trotz ihrer evolutionären Bedeutung sind ihre Genome nach wie vor nur lückenhaft kartiert, insbesondere für Arten, die in afrikanischen Gewässern leben. Fontitrygon garouaensis ist besonders bemerkenswert: Es ist der einzige bekannte Stachelrochen, der vollständig an afrikanische Süßwassersysteme angepasst ist, und es zeigt auffällige Körpermerkmale wie eine reduzierte oder fehlende Reihe von Dornen auf dem Rücken. Da der Übergang vom salzigen Meer in frische Flüsse für Haie und Rochen selten und physiologisch herausfordernd ist, bietet diese Art ein natürliches Experiment dafür, wie Tiere ihre Biologie umbauen, um mit neuen Lebensräumen zurechtzukommen.
Aufbau einer genetischen Referenzkarte
Um den genetischen Bauplan des Rochens zu entschlüsseln, sammelten die Forscher ein einzelnes adultes Weibchen aus dem Niger in Nigeria und isolierten hochwertige DNA und RNA aus mehreren Geweben. Sie verwendeten dann eine „hybride“ Strategie, die drei hochmoderne Sequenzieransätze kombiniert. Kurzlese-Illumina-Sequenzierung liefert enorme Mengen winziger DNA-Fragmente, Langlese-PacBio-HiFi-Sequenzierung erzeugt wesentlich längere und zusammenhängendere Abschnitte, und Hi-C-Sequenzierung erfasst, wie DNA im Zellkern gefaltet ist und interagiert. Durch das Verflechten dieser Datensätze mit spezialisierter Software assemblierte das Team ein Genom von etwa 4,19 Milliarden DNA-Basen, organisiert in 41 Chromosomen-ähnlichen Stücken – vergleichbar in der Größe mit einigen der größten bekannten Hai-Genome.
Was das Genom enthüllt
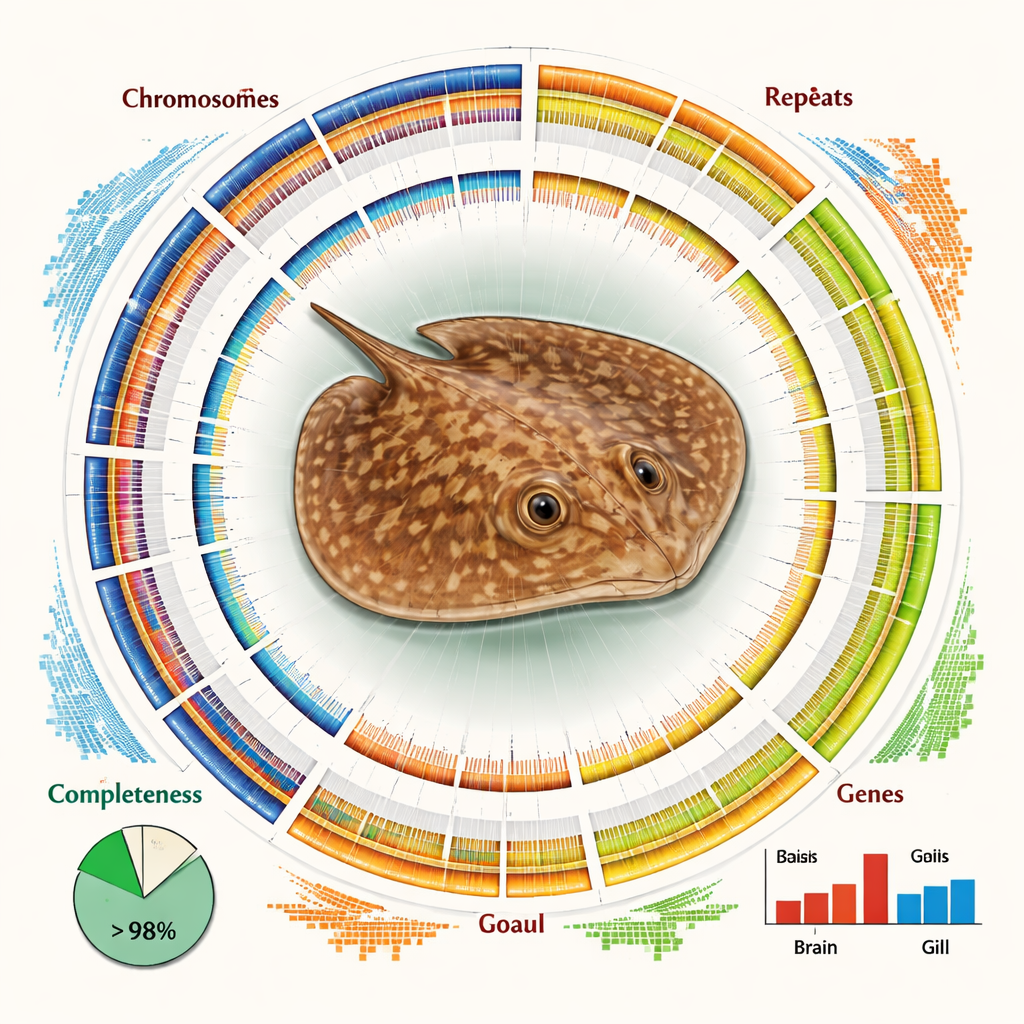
Die fertige Assemblierung ist sowohl groß als auch bemerkenswert vollständig. Unabhängige Qualitätsprüfungen zeigen, dass mehr als 93 Prozent der erwarteten Kerngene der Wirbeltiere vorhanden und intakt sind, und über 84 Prozent der gesamten DNA-Sequenz lassen sich mit hoher Sicherheit bestimmten Chromosomen zuordnen. Das Genom wird von repetitiver DNA dominiert – Sequenzabschnitte, die vielfach vorkommen – und macht ungefähr zwei Drittel seiner Länge aus. Viele dieser Wiederholungen gehören zu mobilen genetischen Elementen wie LINEs und LTRs, die sich im Laufe der Evolution kopiert und im Genom eingefügt haben. Trotz dieser Komplexität identifizierten die Forschenden fast 30.000 proteinkodierende Gene und konnten mithilfe internationaler Protein- und Pfaddatenbanken wahrscheinliche Funktionen für über 98 Prozent von ihnen zuweisen.
Anhaltspunkte für Leben im Süßwasser und Evolution
Eine Chromosomenebenen-Karte ermöglichte dem Team mehr als nur das Auflisten von Genen. Durch den Vergleich der Chromosomen des Rochens mit denen verwandter Haie und Rochen entdeckten sie eine Geschichte struktureller Umgestaltungen – Brüche, Fusionen und Umbauten ganzer Chromosomenabschnitte –, die diese Linie auszeichnen. Diese Veränderungen könnten zur Herausbildung der einzigartigen Merkmale und der ökologischen Nische der Art beigetragen haben. Die Autoren untersuchten außerdem die Genaktivität in zwei Schlüsselgeweben: den Kiemen, die Atmung und Salzhaushalt mit dem umgebenden Wasser vermitteln, und dem Gehirn. Sie fanden deutliche Unterschiede darin, welche Gene aktiviert sind, wobei Kiemengene für Pfade angereichert waren, die mit Energieverbrauch und Fettstoffwechsel zu tun haben – ein Hinweis auf die biochemischen Anpassungen, die nötig sind, um im Süßwasser zu überleben.
Eine neue Grundlage für den Naturschutz
Kern dieser Arbeit ist, dass ein wenig bekannter, bedrohte Flussrochen zu einem der am besten charakterisierten Knorpelfische auf DNA-Ebene wird. Für Nichtfachleute bedeutet das: Wissenschaftler haben jetzt ein detailliertes Referenzmanual für die Art – von Chromosomen bis zu einzelnen Genen. Diese Referenz kann künftige Studien darüber leiten, wie sie mit sich verändernder Wasserqualität umgeht, wie ihre Populationen über Flusssysteme verteilt sind und wie sie mit anderen Rochen verwandt ist. Ebenso wichtig ist, dass sie eine genomische Grundlage für die Naturschutzplanung bietet, neue Möglichkeiten eröffnet, genetische Vielfalt zu überwachen, zu schützende, getrennte Populationen zu identifizieren und zu verstehen, wie dieser seltene Süßwasser-Spezialist auf anhaltende Umweltbelastungen reagieren könnte.
Zitation: Nneji, L.M., Oladipo, S.O., Oyebanji, O.O. et al. First Chromosome-level Genome Assembly and Annotation of an Endangered Freshwater Stingray (Fontitrygon garouaensis) from Africa. Sci Data 13, 302 (2026). https://doi.org/10.1038/s41597-026-06646-0
Schlüsselwörter: Süßwasser-Stachelrochen, Genomassemblierung, Elasmobranch-Evolution, Erhaltungsgenomik, afrikanische Fluss-Biodiversität