Clear Sky Science · de
Verhinderung von Proteomik-Datenfriedhöfen durch kollektive Verantwortung und Community‑Engagement
Warum Ihre medizinischen Daten nicht in einem digitalen Grabhügel enden sollten
Die moderne Medizin stützt sich zunehmend auf riesige Datensätze, die die Tausenden von Proteinen in unseren Zellen beschreiben. Diese Dateien werden oft offen im Internet geteilt, mit dem Versprechen, dass andere Wissenschaftler Ergebnisse überprüfen oder neue Fragen stellen können, ohne neue Experimente durchzuführen. Wenn die Daten jedoch in verwirrenden Formaten veröffentlicht, wichtige Details fehlen oder sie an proprietäre Software gebunden sind, werden sie zu „Datenfriedhöfen“: für alle sichtbar, aber praktisch unbrauchbar. Dieser Artikel beschreibt, wie ein Universitätskurs Studierende zu Daten‑Detektiven machte, um dieses versteckte Problem aufzudecken, und schlägt einfache Verbesserungen vor, die geteilte Daten wirklich wiederverwendbar machen könnten.
Wissenschaft lernen, indem man echte Studien wiederholt
An der Universität Helsinki wurden Doktorandinnen und Doktoranden eines Proteomik‑Kurses in Massenspektrometrie gebeten, etwas Ambitioniertes zu tun: reale, öffentlich verfügbare Proteindatensätze aus einem großen Repository auszuwählen und zu versuchen, die veröffentlichten Befunde zu reproduzieren. In kleinen Gruppen luden sie sechs Projekte aus dem ProteomeXchange‑Netzwerk herunter, das Massenspektrometrie‑Ergebnisse vieler Labore weltweit beherbergt. Mit einer gemeinsamen Analyse‑Pipeline in der Programmiersprache R folgten die Studierenden den gleichen groben Schritten wie die ursprünglichen Forschenden: Proteine identifizieren, ihre Häufigkeit messen, die Daten bereinigen und testen, welche Proteine sich zwischen Bedingungen wie erkranktem und gesundem Gewebe verändern.
Große Versprechen, fehlende Anleitungen
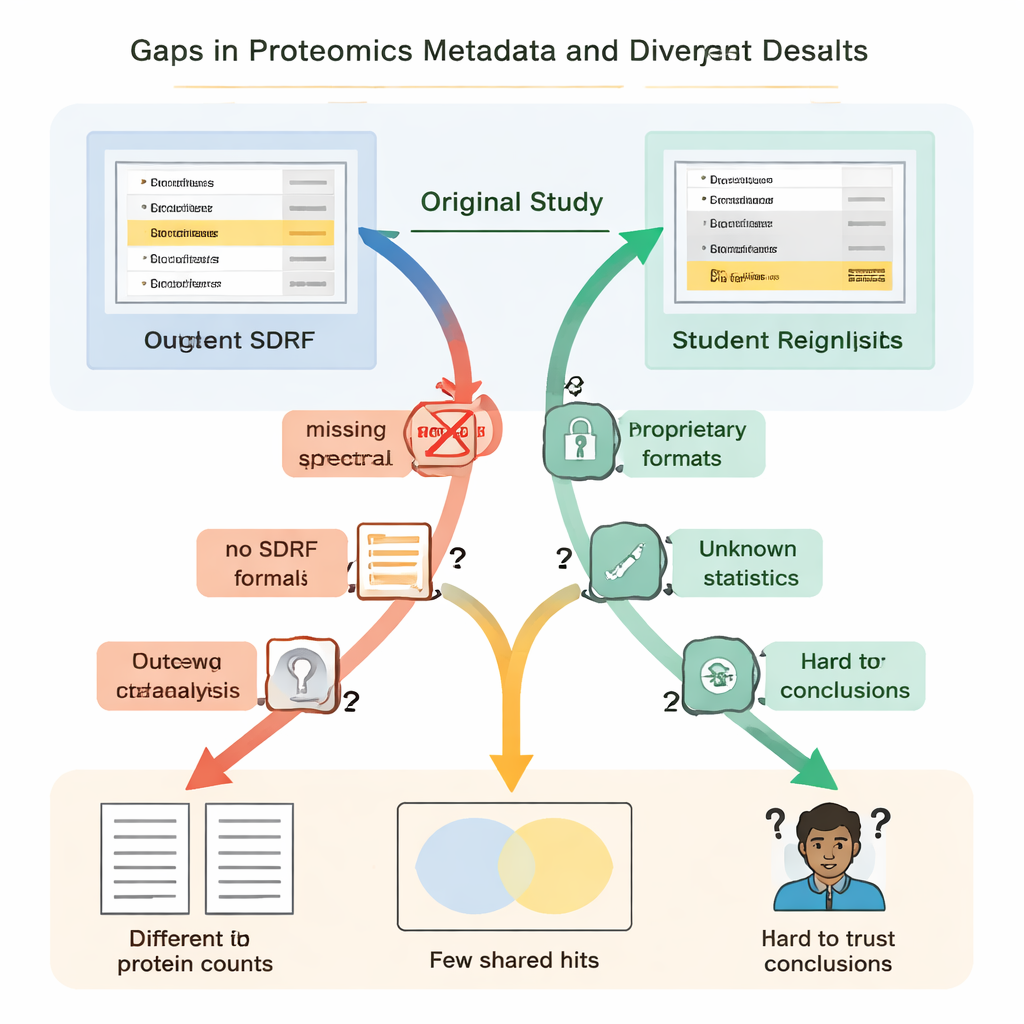
Die Studierenden entdeckten schnell, dass „offen“ nicht immer „wiederverwendbar“ bedeutete. In jedem Fall fehlten wesentliche Anleitungen oder waren schwer zu finden. Wichtige Verknüpfungen zwischen Proben und Datendateien wurden nicht in einem einfachen, maschinenlesbaren Format beschrieben, sodass die Teams anhand von Papiertexten und Dateinamen erraten mussten, welche Rohdateien zu welchen biologischen Gruppen gehörten. Details darüber, wie falsch‑positive Treffer kontrolliert wurden — etwa der Einsatz spezieller „Decoy“-Proteinsequenzen — fehlten, wodurch es unmöglich war, die Vertrauenswürdigkeit der berichteten Proteinlisten rigoros zu beurteilen. In mehreren Projekten waren die Hauptergebnisse in proprietären Dateiformaten gesperrt oder hingen von kommerzieller Software ab, auf die die Studierenden keinen Zugriff hatten, sodass sie große Teile der Analyse von Grund auf neu erstellen mussten.
Wenn kleine Lücken große Unterschiede erzeugen
Diese fehlenden Informationen waren nicht nur lästig; sie führten zu dramatisch unterschiedlichen wissenschaftlichen Ergebnissen. In einer Nierenerkrankungsstudie meldeten die ursprünglichen Autorinnen und Autoren knapp fünftausend Proteine, während die Reanalyse der Studierenden — unter Verwendung eines offenen Werkzeugs und einer selbst erstellten Spektralbibliothek — über dreizehntausend fand. Ein im Originalpapier als besonders wichtig hervorgehobenes Protein tauchte in der zugrunde liegenden Identifikationsdatei nicht überzeugend auf und wurde im Workflow der Studierenden überhaupt nicht nachgewiesen. In einem anderen Fall listete die Originalstudie 108 Proteine als zwischen Bedingungen veränderlich auf, doch die Studierenden konnten, ausgehend von denselben Rohdaten aber ohne vollständige Informationen zur ursprünglichen Statistik, nur 11 sicher identifizieren. An anderer Stelle machte das Fehlen biologischer Replikate in den hochgeladenen Dateien ordentliche statistische Tests schlicht unmöglich.
Was ein „wiederverwendbarer“ Datensatz wirklich enthalten sollte
Aus diesen sechs Fallstudien ergab sich ein klares Muster: Die Hauptbarrieren für Reproduzierbarkeit waren nicht die Massenspektrometer selbst, sondern die Art und Weise, wie Ergebnisse verpackt und geteilt wurden. Die Autorinnen und Autoren argumentieren, dass jeder Proteomik‑Datensatz ein minimales Re‑Analyse‑Paket enthalten sollte. Dazu gehören Rohdaten plus offene, community‑standardisierte Ergebnisformate; eine standardisierte Tabelle, die jede Probe mit ihren experimentellen Bedingungen verknüpft; grundlegende Qualitätskontroll‑Zusammenfassungen; alle Spektralbibliotheken oder Proteinsequenzdateien, die nötig sind, um die Suche zu wiederholen; sowie vollständige Analyseparameter und Code, idealerweise zusammen mit versionierter Software in Containern. Repositorien, Zeitschriften und Gutachter könnten helfen, indem sie Einreichende dazu anregen oder verpflichten, dieses Paket von vornherein bereitzustellen, sodass andere nicht den Workflow aus verstreuten Hinweisen rekonstruieren müssen.
Wissenschaftler ausbilden und gleichzeitig das System reparieren
Der Kurs selbst erfüllte einen doppelten Zweck. Für die Studierenden bot er eine praxisnahe Möglichkeit, komplexe Proteomik‑Methoden, Statistik und Programmierung zu beherrschen, und machte zugleich deutlich, wie fragil veröffentlichte Schlussfolgerungen sein können, wenn die Dokumentation unvollständig ist. Für die breitere Community lieferten die Schwierigkeiten der Studierenden einen Stresstest der aktuellen Praktiken beim Teilen von Daten und zeigten genau auf, wo Metadaten und Analyseprotokolle mangelhaft sind. Die Autorinnen und Autoren schlagen vor, ähnliche Kurse andernorts anzubieten und Klassenzimmer in Qualitätskontroll‑Motoren zu verwandeln, die kontinuierlich auf klarere, transparentere Daten drängen.
Von Datenfriedhöfen zu lebendigen Ressourcen
Kurz gesagt kommen die Verfasser zu dem Schluss, dass viele Proteindatensätze, die in öffentlichen Repositorien liegen, Gefahr laufen, zu digitalen Friedhöfen zu werden — teure Experimente, deren Ergebnisse nicht zuverlässig geprüft oder erweitert werden können. Die Lösung ist jedoch relativ schlicht: Behandeln Sie Metadaten, offene Formate und teilbaren Code als integrale Bestandteile des Experiments, nicht als Nachgedanken. Wenn Forschende, Gutachter und Repositorien gemeinsam auf ein einfaches, gut dokumentiertes Paket bestehen, sobald Proteomik‑Daten geteilt werden, können diese Datensätze „lebendig“ bleiben: bereit zur Reanalyse, kombinierbar mit neuen Studien und nutzbar, um die Evidenzlage in der biomedizinischen Forschung zu stärken.
Zitation: Vadadokhau, U., Soliman, M., Castillon, L. et al. Preventing Proteomics Data Tombs Through Collective Responsibility and Community Engagement. Sci Data 13, 287 (2026). https://doi.org/10.1038/s41597-026-06614-8
Schlüsselwörter: Proteomik, Datenreproduzierbarkeit, Open Science, Massenspektrometrie, Forschungsdatenfreigabe