Clear Sky Science · de
AF2BIND: Vorhersage von Bindungsstellen für kleine Moleküle mithilfe der Paar-Repräsentation von AlphaFold2
Wirkstoffziele finden in einem Meer von Proteinen
Moderne Medikamente wirken häufig, indem sie an winzige Nischen und Vertiefungen auf der Oberfläche von Proteinen in unseren Zellen andocken. Selbst mit den heute verfügbaren umfangreichen Katalogen von Proteinstrukturen ist es jedoch überraschend schwierig vorherzusagen, wo ein kleines Molekül – ein potenzielles Medikament – tatsächlich haften könnte. Diese Studie stellt AF2BIND vor, ein einfaches, aber wirkungsvolles rechnerisches Werkzeug, das die internen Daten von AlphaFold2, dem bahnbrechenden Prädiktor von Proteinstrukturen, auswertet, um wahrscheinliche Wirkstoffbindungsstellen über tausende menschliche Proteine hinweg hervorzuheben. Ziel ist es, die Suche nach neuen Medikamenten zu fokussieren und verborgene funktionelle Hotspots aufzudecken, die traditionelle Methoden übersehen.
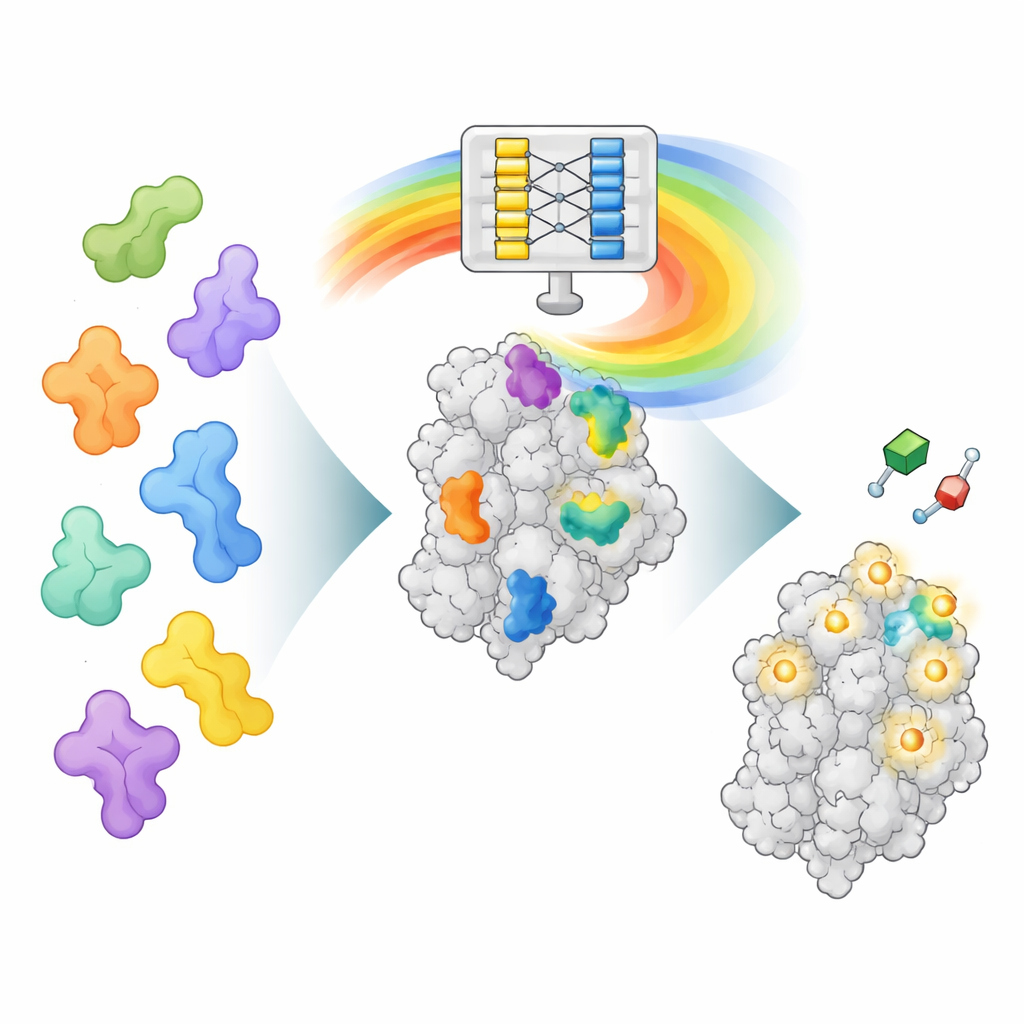
Eine neue Art, AlphaFolds „Denken“ zu lesen
AlphaFold2 wurde darauf trainiert vorherzusagen, wie sich eine Aminosäurekette in ein dreidimensionales Protein faltet, nicht darauf, wo Medikamente binden. Beim Lernen des Faltens hat es jedoch auch reichhaltige Muster darüber erworben, wie verschiedene Teile von Proteinen miteinander interagieren. AF2BIND zapft eine dieser internen Datenebenen an, die sogenannte Paar-Repräsentation, welche kodiert, wie jedes Aminosäurepositionspaar räumlich zueinander steht. Die Autoren füttern AlphaFold2 mit einer Proteinsequenz zusammen mit ihrer Rückgratstruktur und hängen zusätzlich 20 extra Aminosäuren an, je eine pro Typ, als separate „Köder“-Ketten. AlphaFold2 berechnet dann, wie das Protein mit jeder Köderrest-Position interagiert. Diese Interaktionsmuster werden Eingabe für ein sehr geradliniges logistisches Regressionsmodell, das für jede Position im Protein die Wahrscheinlichkeit schätzt, dass sie zu einer Bindungsstelle für kleine Moleküle gehört.
Verborgene Signale in praktische Vorhersagen verwandeln
Das Training von AF2BIND erforderte einen sorgfältig kuratierten Satz von etwa 1.900 Protein–Ligand-Strukturen, bei denen kleine Moleküle mit hochwertigen experimentellen Nachweisen gebunden sind. Die Forschenden unternahmen erhebliche Anstrengungen, um „Schummeln“ durch Ähnlichkeit zu vermeiden: Sie teilten ihre Daten so auf, dass Testproteine weder die Gesamttopologie, noch die Sequenz oder sogar die Form der Bindungstasche mit den Trainingsdaten teilten. Auf diesem strengen Benchmark übertraf die AF2-Paar-Repräsentation mehrere alternative neuronale Einbettungen, einschließlich solcher, die nur auf Sequenz oder auf struktur-konditioniertem Sequenzdesign basieren. Mit den Paarmerkmalen allein fand AF2BIND etwa zwei Drittel der bekannten Bindungsreste in den bestbewerteten Vorhersagen wieder und zeigte starke Leistung über gängige Klassifikationsmetriken hinweg, während es robust gegenüber moderaten Änderungen in Proteinfaltung und Seitenkettenorientierung blieb.
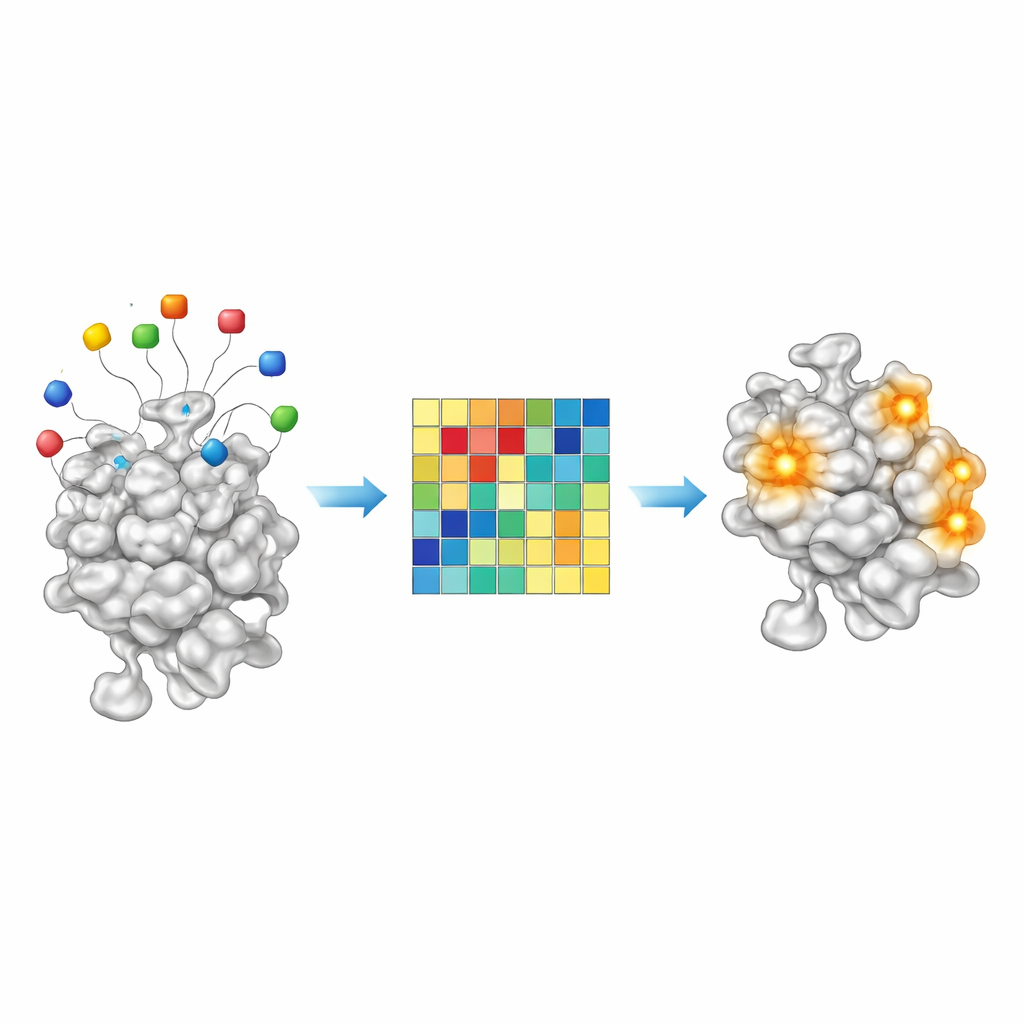
Chemische Hinweise aus Köderresten lesen
Da AF2BIND ein einfaches lineares Modell ist, sind seine Entscheidungen für ein modernes KI-System ungewöhnlich transparent. Jeder der 20 Köder-Aminosäuren leistet einen messbaren Beitrag zur finalen Bindungsbewertung an einer gegebenen Proteinposition. Durch die Untersuchung dieser Beiträge über rund 2.000 Protein–Ligand-Komplexe hinweg stellten die Autoren fest, dass bestimmte Köderkombinationen stärker ansprechen bei öligen, kohlenstoffreichen Liganden, während andere bei polareren, wasserliebenden Molekülen aufleuchten. Anders ausgedrückt fungiert das Muster der Köderaktivierung wie ein grober chemischer Fingerabdruck dafür, welche Arten von kleinen Molekülen eine bestimmte Tasche bevorzugt. Das deutet darauf hin, dass AF2BIND-ähnliche Ansätze künftig nicht nur markieren könnten, wo ein Wirkstoff binden kann, sondern auch Hinweise auf die passende Chemie liefern könnten.
Das menschliche Proteom nach neuen Taschen absuchen
Mit ihrem trainierten Modell setzten die Forschenden AF2BIND auf den AlphaFold-vorhergesagten Strukturen des gesamten menschlichen Proteoms ein. Nach dem Entfernen von Regionen mit geringer Vertrauenswürdigkeit und dem Aufteilen sehr großer Proteine in handhabbare Strukturbereiche gruppierten sie nahe beieinanderliegende hochbewertete Reste zu Kandidaten-Bindungsstellen. AF2BIND sagte über 20.000 solcher Stellen in mehr als 13.000 Proteinen voraus. Bemerkenswert ist, dass die Mehrheit dieser Stellen nicht mit Taschen übereinstimmte, die durch homologiebasierte Methoden wie AlphaFill – die Liganden aus verwandten Kristallstrukturen übernimmt – abgeleitet wurden, noch mit einem weit verbreiteten Taschenfinder namens P2Rank. Viele nur von AF2BIND gefundene Stellen sind flacher oder diffuser als klassische vergrabene Taschen und fallen oft mit Regionen zusammen, die Peptide, RNA, DNA oder andere Proteine binden – Schnittstellen, die dennoch durch kleine Moleküle angreifbar sein könnten.
Folgen für Arzneimittelentdeckung und Krankheit
Um einzuschätzen, wie vielversprechend diese neu vorgeschlagenen Stellen für die Wirkstoffentwicklung sind, verwendeten die Autoren ein unabhängiges Werkzeug, das die „Druggability“ anhand von Taschengröße, Einkapselung und chemischer Umgebung bewertet. Im Durchschnitt lagen AF2BINDs Stellen über einem gebräuchlichen Schwellenwert für attraktive Wirkstoffziele, einschließlich solcher in Proteinen, die mit vererbten Krankheiten in Verbindung stehen. Beim Abgleich mit chemoproteomischen Experimenten, die reaktive Cysteine in Zellen markieren, erklärten AF2BIND und P2Rank zusammen nahezu die Hälfte der beobachteten ligandierbaren Regionen, wobei jede Methode Fälle erfasste, die die andere verfehlte. Die Arbeit zeigt, dass die internen Repräsentationen von Strukturvorhersage-Netzen umfunktioniert werden können, um wahrscheinlichte Wirkstoffbindungsstellen in großem Maßstab zu kartieren, ohne Vorwissen über ein bestimmtes Ligand. Für Nichtfachleute lautet die Kernbotschaft: Dieselben KI-Durchbrüche, die Proteinformen vorhersagen, beginnen aufzuzeigen, wo und wie Medikamente am besten an diesen Formen andocken könnten, was die Suche nach neuen Therapien beschleunigen und bislang verborgene Kontrollpunkte in unseren Proteinen beleuchten könnte.
Zitation: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Schlüsselwörter: Protein-Bindungsstellen, Arzneimittelentdeckung, AlphaFold2, Computationale Biologie, Strukturelle Bioinformatik