Clear Sky Science · de
DECODE: Deep‑Learning‑basiertes, gemeinsames Deconvolution‑Framework für verschiedene Omics‑Daten
Warum diese Forschung wichtig ist
Die moderne Biomedizin ist überschwemmt mit Messungen unserer Gewebe: welche Gene aktiv sind, welche Proteine vorhanden sind und welche kleinen Moleküle unsere Zellen antreiben. Die meisten dieser Messungen stammen jedoch aus gemischten Proben, in denen zahlreiche Zelltypen zusammen vorliegen. Die Studie zu DECODE stellt ein leistungsfähiges KI‑Framework vor, das diese Signale entmischen kann und so aufzeigt, welche Zellen und Zellzustände vorhanden sind – selbst über sehr unterschiedliche Datentypen hinweg. Diese Fähigkeit kann die Forschung zu Krebs, Immunologie und Stoffwechselerkrankungen beschleunigen und den Wert bereits vorhandener Biobankproben erhöhen.
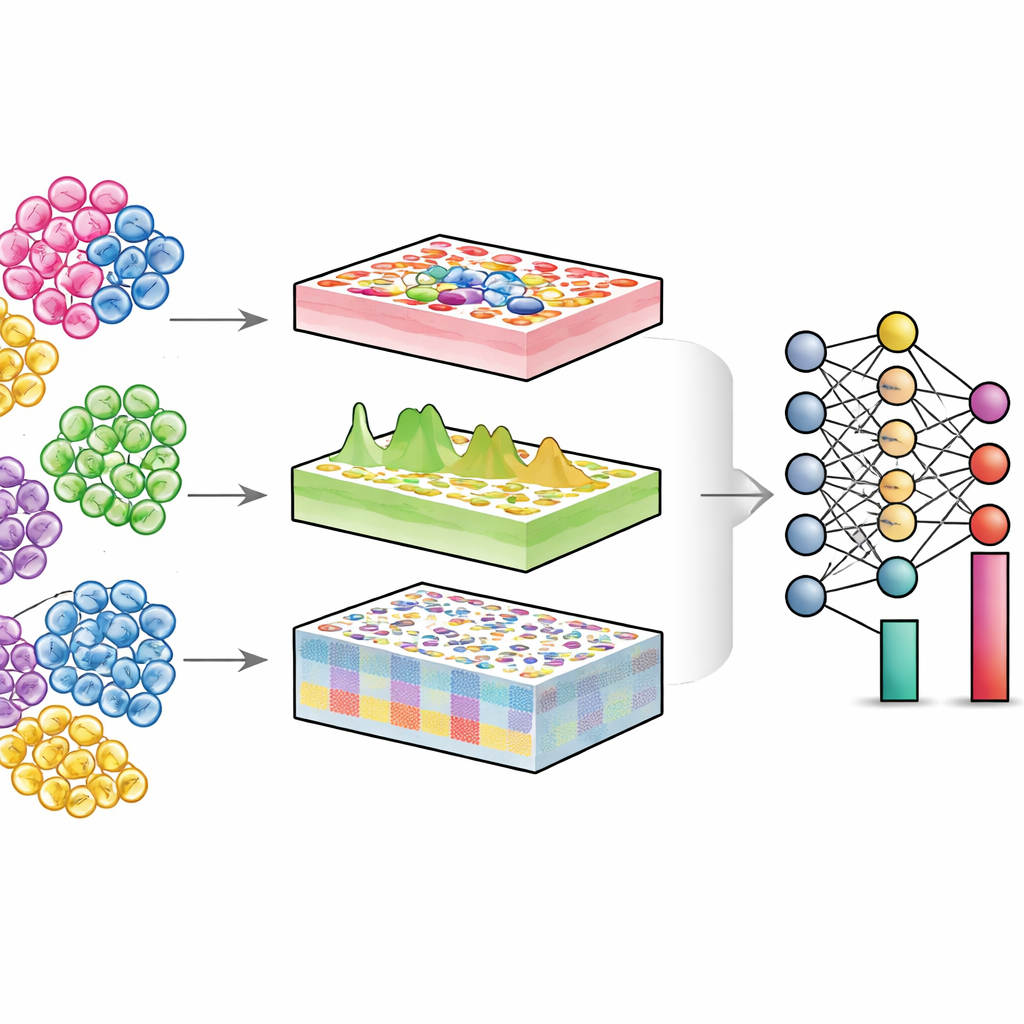
Ein Blick in gemischte Gewebe
Jedes Organ ist eine Gemeinschaft unterschiedlicher Zelltypen – Immunzellen, strukturelle Zellen, Stammzellen und mehr. In Gesundheit und Krankheit ändert sich oft nicht nur die Funktion einzelner Zellen, sondern auch deren Häufigkeit und ihr Zustand. Single‑Cell‑Technologien messen einzelne Zellen direkt, sind aber teuer und technisch anspruchsvoll, besonders bei großen Patientenkohorten oder älteren, gelagerten Proben. Konventionelle „Bulk“‑Experimente hingegen mischen Tausende bis Millionen von Zellen und liefern ein gemitteltes Signal. Deconvolution‑Algorithmen versuchen, diese Mischung umzukehren: Aus Bulk‑Daten und einer Single‑Cell‑Referenz schätzen sie den Anteil der einzelnen Zelltypen im Gewebe.
Die Grenzen spezialisierter Werkzeuge
Bestehende Deconvolution‑Werkzeuge sind meist auf einen einzelnen Messtyp zugeschnitten, etwa Genaktivität (Transkriptomik) oder Proteine (Proteomik). Sie gehen oft von statistischen Annahmen aus, die für andere Datentypen nicht gelten, und stoßen an Grenzen, wenn das Bulk‑Gewebe Zelltypen enthält, die in der Referenz fehlen. Starke Batch‑Effekte – Unterschiede zwischen Spendern, Geräten oder Gesundheitszuständen – können die biologischen Signale zusätzlich verwässern. Bemerkenswert ist, dass es zuvor keine praktikable Methode für die Metabolomik gab, die die kleinen Moleküle untersucht, die klinischen Symptomen oft am nächsten stehen. Daher mussten Forscher mit Multiomics‑Kohorten mehrere spezialisierte Werkzeuge verwenden, jedes mit eigenen Eigenheiten, was Vergleiche zwischen Studien und Datentypen erschwerte.
Eine universelle Entmischungs‑Engine
DECODE begegnet diesen Herausforderungen, indem es Deconvolution als flexibles Deep‑Learning‑Problem formuliert, das Gene, Proteine und Metaboliten einheitlich verarbeiten kann. Zunächst synthetisiert es „Pseudogewebe“, indem Single‑Cell‑Profile in zufälligen Anteilen digital gemischt werden und so einen umfangreichen Trainingssatz mit bekannter Zellzusammensetzung entsteht. Eine adversarielle Lernphase lehrt dann einen Encoder, reale Gewebe und Pseudogewebe in eine gemeinsame Repräsentation zu überführen, in der technische Unterschiede minimiert, biologisch relevante Muster jedoch erhalten bleiben. Anschließend lernt ein spezielles Denoising‑Modul, geleitet durch contrastives Lernen, wahre Gewebesignale von künstlichem Rauschen zu trennen. Dieser Schritt macht DECODE robust gegenüber fehlenden Zelltypen in der Referenz und gegenüber Messfehlern. Schließlich werden die bereinigten Merkmale an ein Deconvolution‑Modul übergeben, das je nach Vollständigkeit der Referenz absolute oder relative Häufigkeiten von Zelltypen und Zellzuständen schätzt.
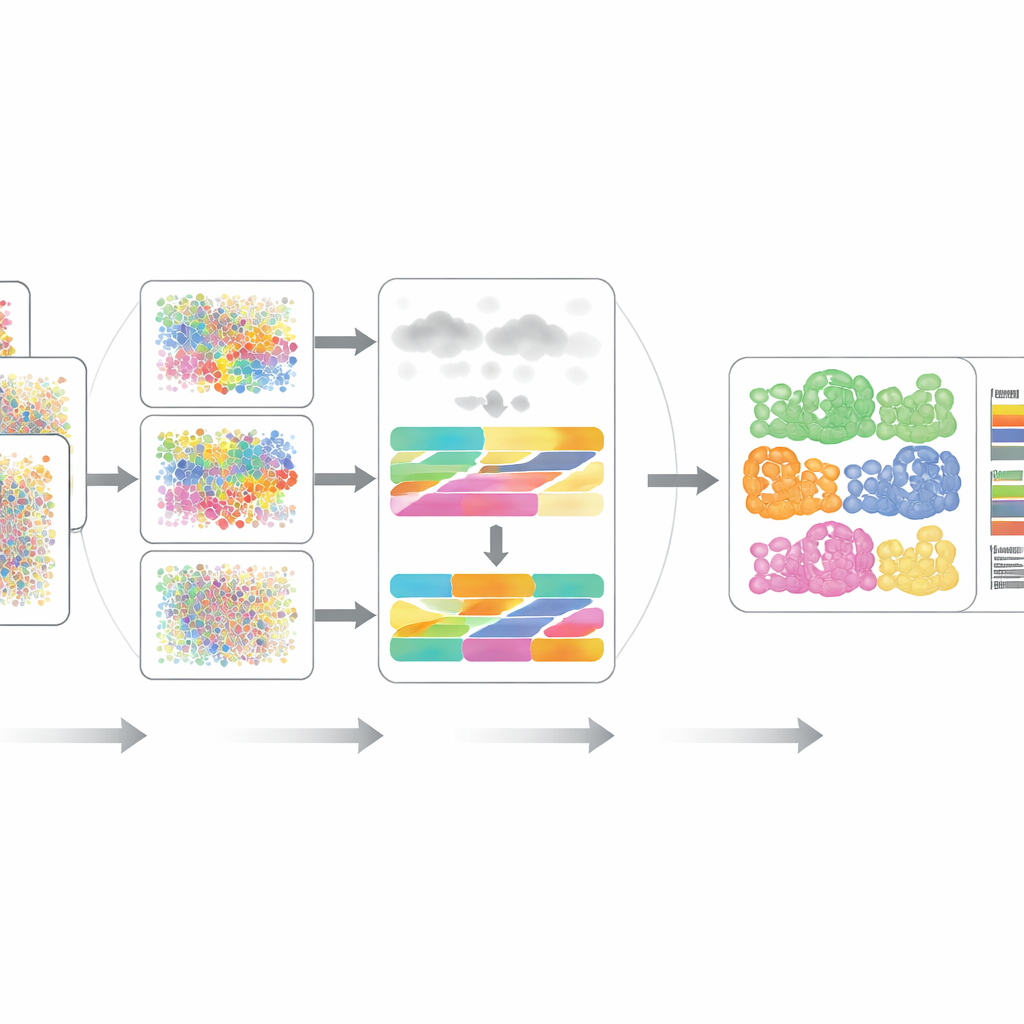
DECODE auf dem Prüfstand
Die Autoren evaluierten DECODE rigoros an 15 Datensätzen, die sieben realistische Szenarien abdeckten, darunter unterschiedliche Spender, Krankheitszustände, Gesundheitsbedingungen, experimentelle Plattformen und sogar räumlich aufgelöste Messungen. In Transkriptomik und Proteomik erreichte DECODE im Allgemeinen die Genauigkeit moderner Methoden und übertraf diese teilweise, bei gleichzeitig angemessenem Rechen‑ und Speicheraufwand. Entscheidenderweise war DECODE die einzige Methode, die verlässliche Ergebnisse für Metabolomik‑Daten lieferte, in denen es weniger Merkmale gibt und verschiedene Zelltypen täuschend ähnlich aussehen können. Das Framework erwies sich zudem als fähig, Zellzustände nachzuverfolgen – etwa Entwicklungsverläufe, Zellzyklus‑Phasen oder Reaktionen auf Medikamentenbehandlung – und nicht nur statische Zelltypen.
Robust gegenüber verrauschten und unvollständigen Real‑Daten
Reale Gewebe enthalten oft Zelltypen, die in Labor‑basierten Single‑Cell‑Referenzen nicht erfasst sind, und experimentelles Rauschen kann viele Merkmale gleichzeitig verzerren. Die Forscher simulierten diese Probleme, indem sie unbekannte Zelltypen hinzufügten und verschiedene Arten von Rauschen und fehlenden Daten in Transkriptomik, Proteomik und Metabolomik einführten. In den meisten Szenarien blieb DECODE die genaueste Methode und in der Metabolomik die einzige, die nicht zusammenbrach. Außerdem zeigten sie, dass DECODE bei passenden Gen‑ und Proteinmessungen aus denselben Blutproben sehr konsistente Antworten liefert – eine wichtige Voraussetzung, um Zelltypveränderungen über Omics‑Ebenen in großen Kohorten zu vergleichen.
Neue biologische Einsichten aus Multiomics‑Kohorten
Mit diesem einheitlichen Werkzeug untersuchten die Autoren komplexe Krankheitsdatensätze neu. Beim Brustkrebs kombinierten sie transkriptomische und proteomische Kohorten, um zu zeigen, wie sich Immunzellen und unterstützende Stromazellen zwischen nicht‑metastatischen Tumoren, metastasierenden Primärtumoren und Hirnmetastasen verschieben. Muster wie höhere T‑Zell‑ und perivaskulär‑ähnliche Zellvorkommen in nicht‑metastatischen Läsionen und vermehrte B‑Zellen in fortgeschrittener Erkrankung stimmen mit früheren Studien überein und ergänzen diese. In der Mausleber integrierte DECODE transkriptomische, proteomische und metabolomische Kohorten, um nachzuverfolgen, wie Hepatozyten, Endothelzellen und residente Immunzellen sich bei unterschiedlichen Diäten und Lebererkrankungsmodellen verändern, und reproduzierte bekannte Trends wie steigende Kupffer‑Zell‑Anteile bei entzündlichen Zuständen.
Was das für die Zukunft bedeutet
Für eine allgemeine Leserschaft lautet die Kernbotschaft: DECODE wirkt wie ein intelligentes Prisma für biomedizinische Daten. Anhand gemischter Gewebemessungen kann es die Beiträge vieler verschiedener Zelltypen und ‑zustände trennen und dies zuverlässig über mehrere molekulare Messarten hinweg. Dadurch können Wissenschaftler deutlich mehr Informationen aus bestehenden Multiomics‑Kohorten und Biobanken gewinnen, ohne für jedes Projekt neue Single‑Cell‑Daten erheben zu müssen. Zwar hängt die Methode weiterhin von Qualität und Umfang verfügbarer Single‑Cell‑Referenzen ab und Metabolomik‑Ressourcen sind begrenzt, doch markiert DECODE einen wichtigen Schritt hin zu einer routinemäßigen, zellbasierten Interpretation groß angelegter Humanstudien mit potenziellen Vorteilen für das Verständnis von Krankheitsmechanismen und die Präzisionsmedizin.
Zitation: Zhao, T., Liu, R., Sun, Y. et al. DECODE: deep learning-based common deconvolution framework for various omics data. Nat Methods 23, 596–608 (2026). https://doi.org/10.1038/s41592-026-03007-y
Schlüsselwörter: Multiomics‑Deconvolution, Single‑Cell‑Referenz, Deep Learning in der Biologie, Metabolomik‑Analyse, Zusammensetzung von Zelltypen