Clear Sky Science · de
Variabilität der Evolutionsrate und wiederkehrende Sequenzfehler in pandemie‑großer Phylogenetik
Warum das für künftige Ausbrüche wichtig ist
Wenn sich ein neues Virus weltweit ausbreitet, eilen Wissenschaftler, seinen genetischen Code zu lesen und seinen Stammbaum zu rekonstruieren. Solche Bäume helfen nachzuverfolgen, wie Varianten entstehen, wie schnell sie sich ausbreiten und ob Kontrollmaßnahmen wirken. Während COVID‑19 jedoch Sequenzierlabore Millionen von SARS‑CoV‑2‑Genomen so schnell erzeugten, begannen versteckte Fehler und Besonderheiten in den Daten das Bild zu verzerren. Diese Arbeit stellt neue Methoden vor, um solch gewaltige genetische Datensätze zu bereinigen und zu interpretieren, und liefert so klarere Einblicke, wie ein pandemisches Virus wirklich evolviert und sich durch Populationen bewegt.
Die Herausforderung: Millionen Genome verständlich machen
Die genomische Epidemiologie verwandelt Virusgenome in praktische Informationen für Entscheidungen des öffentlichen Gesundheitswesens. Für SARS‑CoV‑2 wurden weltweit mehr als 20 Millionen Genome geteilt. Traditionelle evolutionäre Werkzeuge wurden für überschaubarere Probleme entwickelt, etwa für den Vergleich von Genen zwischen Arten, nicht für Millionen nahezu identischer Virussequenzen, die in Echtzeit eintreffen. Auf dieser Skala werden zwei Probleme besonders problematisch. Erstens mutieren einige Stellen im Virusgenom viel häufiger als andere, was nicht verwandte Viren merkwürdig ähnlich aussehen lassen kann. Zweitens können wiederkehrende technische Fehler beim Sequenzieren und in der Datenverarbeitung echte Mutationen imitieren. Beide Effekte erzeugen „falsche Echos“ im Evolutionsbaum und schaffen Unsicherheit darüber, welchen Ästen und Gruppierungen man vertrauen kann.
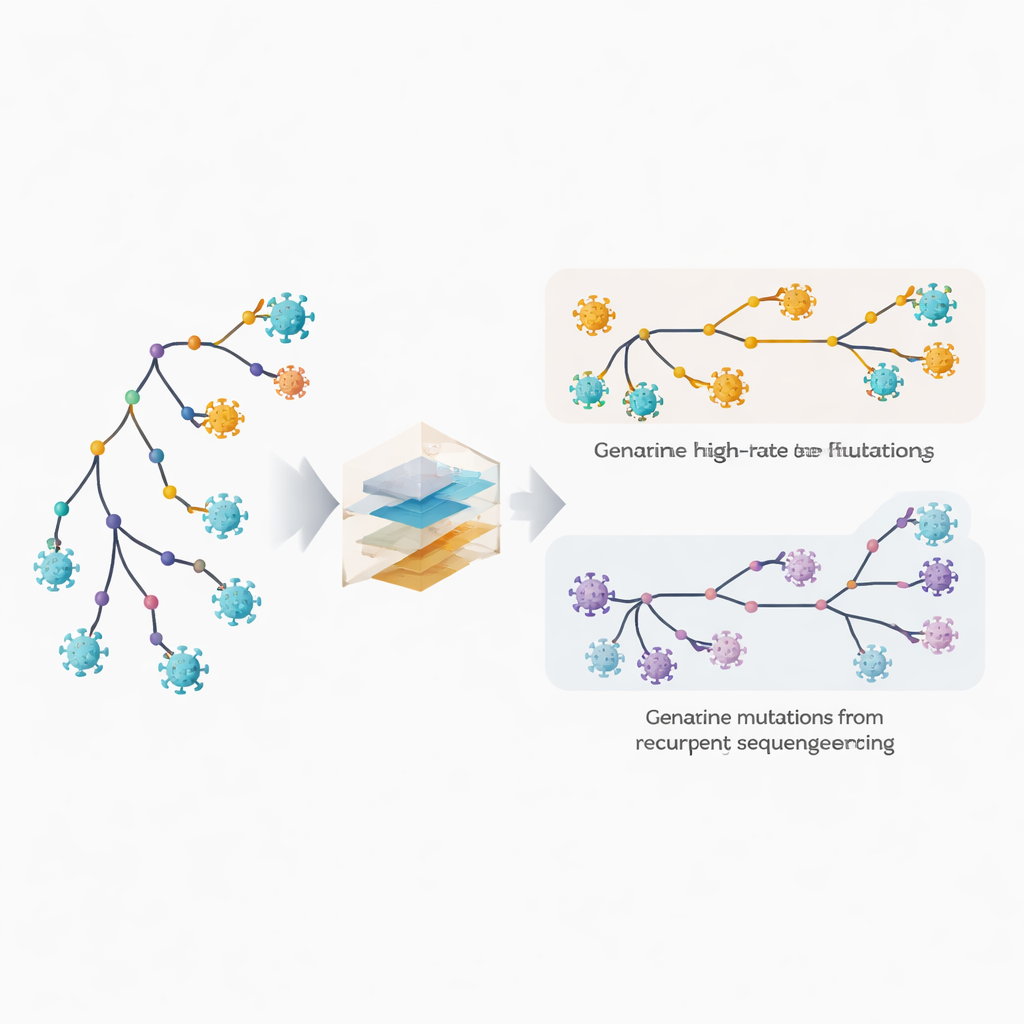
Schnell veränderliche Stellen und verborgene Fehler erkennen
Die Autorinnen und Autoren erweitern ihre phylogenetische Software MAPLE um Modelle, die jede Position im Virusgenom als individuell verhaltend behandeln. Statt einiger weniger durchschnittlicher Mutationsraten schätzt die Methode für jede Position eine eigene Rate, wobei sie die enorme Anzahl verfügbarer Genome ausnutzt. Gleichzeitig erlaubt sie jeder Position eine eigene Wahrscheinlichkeit, wiederkehrende Sequenzier‑ oder Konsensus‑Fehler zu tragen. Der entscheidende Trick besteht darin, zu vergleichen, wie häufig eine Änderung an tiefen inneren Ästen des Baums auftritt, die ältere, geteilte Ereignisse widerspiegeln, gegenüber den äußersten Spitzen, die einzelnen Genomen entsprechen. Echte biologische Mutationen verteilen sich tendenziell zwischen inneren und terminalen Ästen, während technische Fehler überwiegend an den Spitzen erscheinen. Durch Ausnutzen dieses Musters kann die Methode echte schnelle Evolution von wiederholten Fehlern entwirren.
Schnellere Algorithmen für einen überfüllten Baum des Lebens
Das Verarbeiten von Millionen Genomen würde normalerweise enorme Rechenressourcen erfordern. Um die Analyse praktikabel zu halten, hat das Team die Art und Weise neu gestaltet, wie MAPLE Sequenzinformationen auf dem Baum speichert und aktualisiert. Statt jedes Genom mit einer einzelnen festen Referenz zu vergleichen, wählt die Software „lokale Referenzpunkte“ innerhalb des Baums und erfasst nahegelegene Genome als Differenzen relativ zu diesen Ankern. Diese kompakte Darstellung beschleunigt Vergleiche zwischen weit auseinanderliegenden Teilen des Baums. Zusätzliche Verbesserungen verfeinern, wie neue Proben in einen bestehenden Baum eingefügt werden, wie Astlängen angepasst werden und wie wahrscheinliche alternative Baumformen erkundet werden, mit Optionen, die rechenintensivsten Schritte parallel über mehrere Prozessorkerne laufen zu lassen.
Validierung der Methode und Bereinigung realer Daten
Um zu prüfen, ob ihre Modelle funktionieren, erzeugten die Autoren zunächst realistische simulierte SARS‑CoV‑2‑Datensätze mit bekannten Mutationsmustern und eingebetteten Sequenzfehlern. In diesen Tests rekonstruierte der neue Ansatz treuere Evolutionsbäume und lokalisierte einzelne Fehler mit hoher Präzision, insbesondere wenn Zehntausende Genome oder mehr einbezogen wurden. Anschließend wandten sie die Methode auf reale Daten an und analysierten Millionen von SARS‑CoV‑2‑Sequenzen, für die rohe Reads verfügbar waren. Durch den Vergleich zweier verschiedener Konsensus‑Pipelines identifizierten sie spezifische Genompositionen, die wiederholt von Artefakten betroffen waren, etwa durch Primer‑Bindungsprobleme oder referenzverzerrte Aufrufe. Diese verdächtigen Stellen wurden für die weitere Analyse maskiert, und Genome mit Anzeichen von Kontamination oder gemischter Infektion wurden herausgefiltert, wodurch eine kuratierte Alignierung von über zwei Millionen hochwertigen Sequenzen entstand.
Ein klareres globales Bild des Virusstammbaums
Mit dem bereinigten Datensatz rekonstruierten die Autorinnen und Autoren einen globalen SARS‑CoV‑2‑Phylogeniebaum und kartierten, wie große Varianten zueinander in Beziehung stehen. Ihr Baum schlägt manchmal subtil andere Beziehungen vor als frühere öffentliche Bäume, oft in Formen, die weniger Mutationsereignisse benötigen und besser zum statistischen Modell passen. Das Framework hebt außerdem Stellen hervor, an denen Linienbezeichnungen möglicherweise nicht mit der zugrunde liegenden genetischen Geschichte übereinstimmen, und markiert mögliche Rekombinanten oder problematische Genome zur genaueren Überprüfung. Obwohl einige Herausforderungen bestehen bleiben — etwa Überanpassung bei knappen Daten oder der Einfluss stark kontaminierter Proben — zeigt die Arbeit, dass es nun machbar ist, zuverlässigere, pandemie‑große Evolutionsbäume zu erstellen. Für eine allgemeine Leserschaft ist die Quintessenz: Besserer Umgang mit Fehlern und Mutations‑Hotspots führt zu schärferen Einsichten darüber, wie Erreger sich ausbreiten und verändern, und hilft Wissenschaftlern und Gesundheitsbehörden, in künftigen Ausbrüchen schneller und zuversichtlicher zu reagieren.
Zitation: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Schlüsselwörter: SARS‑CoV‑2‑Genomik, phylogenetische Methoden, Sequenzierfehler, Variation der Mutationsrate, genomische Epidemiologie