Clear Sky Science · de
Übertragbare enantioselektivitätsmodelle aus spärlichen Daten
Ein schlauerer Weg, den richtigen Katalysator zu finden
Chemiker suchen oft nach besseren Arzneimitteln und Materialien, indem sie Kohlenstoffatome in sehr spezifischen dreidimensionalen Anordnungen zusammenfügen. Das Erreichen dieses feinen „rechts‑“ gegenüber „links‑“ gerichteten Ergebnisses — bekannt als Enantioselektivität — bedeutet meist, viele Metallkatalysatoren und Reaktionsbedingungen durch Ausprobieren zu testen. Diese Arbeit stellt eine Methode vor, die mit vergleichsweise kleinen Mengen experimenteller Daten kombiniert mit schnellen Computerberechnungen vorhersagt, welche nickelbasierten Katalysatoren in einer Vielzahl von Reaktionen die gewünschte Händigkeit liefern, und so Chemikern womöglich Wochen oder Monate Laborarbeit spart.
Warum Händigkeit von Molekülen so schwer zu kontrollieren ist
Viele Arzneimittel und Naturstoffe existieren als spiegelbildliche Formen, die sich im Körper sehr unterschiedlich verhalten können. Katalysatoren, die eine Spiegelbildform gegenüber der anderen bevorzugen, sind daher extrem wertvoll. Die Entwicklung solcher Katalysatoren ist jedoch anspruchsvoll. Die traditionelle Quantenchemie kann prinzipiell berechnen, welchen Weg eine Reaktion bevorzugt, doch kleinste Energiefehler führen zu großen Fehlern in der vorhergesagten Selektivität, und die Rechnungen sind langsam. Einfachere statistische Modelle sind dagegen schnell, vernachlässigen aber oft die detaillierte Wechselwirkung zwischen Metallkatalysator und reaktiven Molekülen — insbesondere wenn sich der Reaktionsmechanismus je nach eingesetzten Partnern subtil ändert.
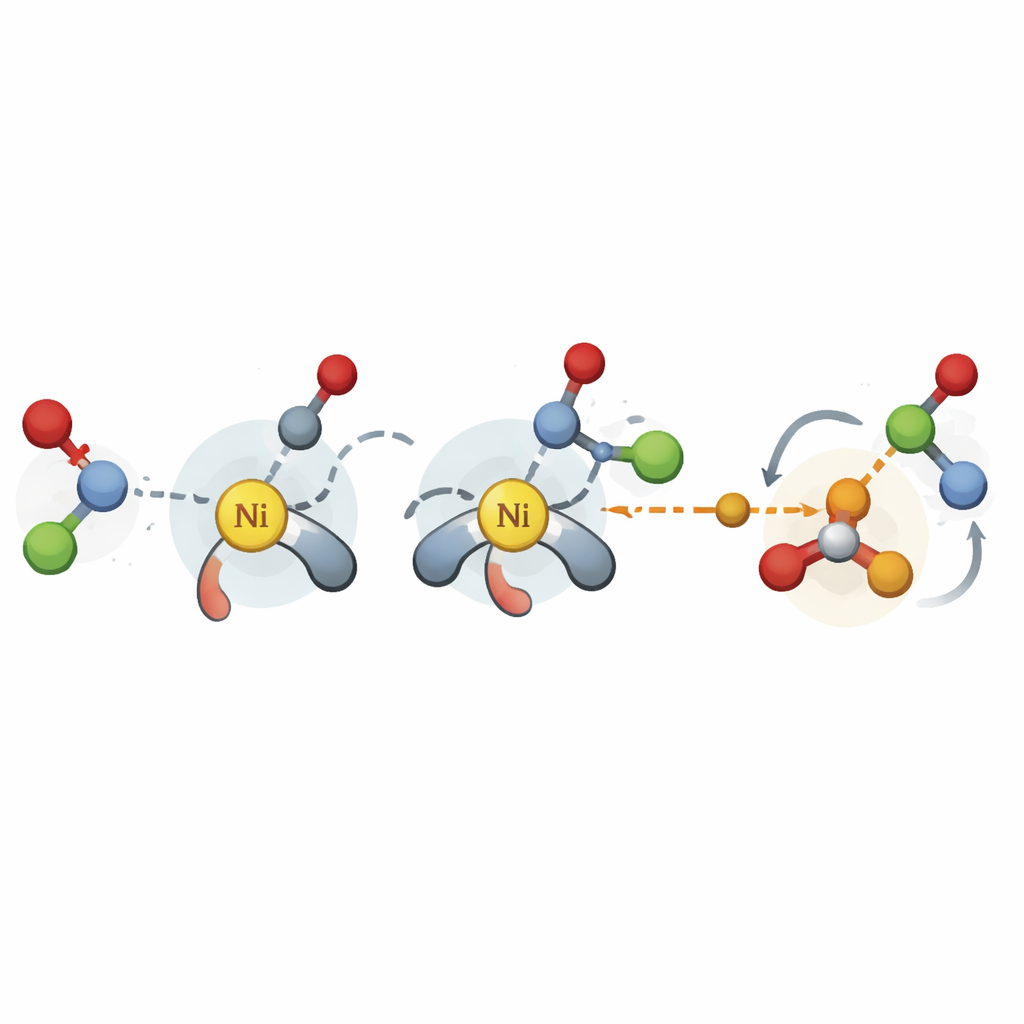
Die wichtigen Momente einer Reaktion erfassen
Die Autoren überbrücken diese Lücke, indem sie sich auf die kritischsten Stadien einer nickelkatalysierten Cross‑Coupling‑Reaktion konzentrieren: die Schritte, in denen neue Kohlenstoff–Kohlenstoff‑Bindungen gebildet werden und das Endprodukt freigesetzt wird. Statt teure hochaufgelöste Simulationen durchzuführen, verwenden sie eine vereinfachte quantenchemische Methode, um dreidimensionale Strukturen für wichtige Übergangszustände und Intermediate über viele mögliche Katalysator‑ und Substratkombinationen hinweg zu erzeugen. Aus diesen Strukturen extrahieren sie Hunderte physikalisch sinnvoller Deskriptoren, etwa wie eng die katalytische Umgebung an bestimmten Atomen ist oder wie leicht sich Elektronen bewegen können. Diese Zahlen fließen dann in einfache lineare Regressionsmodelle, die Strukturmerkmale mit der gemessenen Selektivität in Verbindung bringen.
Aus spärlichen Daten lernen, um neue Experimente zu leiten
Ein zentrales Ergebnis der Arbeit ist, dass sie das Beste aus spärlichen Daten herausholt — den begrenzten Kombinationen von Katalysatoren und Substraten, die typischerweise in einer Publikation berichtet werden. In einer Fallstudie überarbeiten die Autoren eine Nickelreaktion, die Styroloxide mit Aryl‑iodiden koppelt. Sie zeigen, dass Deskriptoren aus dem relevantesten Übergangszustand besser abschneiden als solche aus vereinfachten Katalysatorfragmenten, obwohl die zugrundeliegenden Berechnungen kostengünstiger sind. Mit diesen Modellen testen sie virtuell viele weitere Liganden an vorhandenen Substratpaaren und identifizieren neue Katalysatoroptionen, die den enantiomeren Überschuss für besonders hartnäckige Beispiele erhöhen — und das, ohne Dutzende unnötiger Experimente durchzuführen.
Wissen zwischen verschiedenen Reaktionen übertragen
Die Methode ist mächtig, weil sie auf unterschiedliche, aber verwandte nickelkatalysierte Reaktionen übertragbar ist. In einer zweiten Studienreihe kombinieren die Autoren Daten aus mehreren Typen von Nickelreaktionen, die alle Bindungen zwischen sp3‑hybridisierten Kohlenstoffatomen und Partnern wie aryl‑ oder alkenyl‑Gruppen bilden, selbst wenn sich die genauen Bedingungen oder Kupplungspartner unterscheiden. Indem sie Modelle aus den gleichen mechanistisch sinnvollen Deskriptoren aufbauen, sagen sie erfolgreich die Enantioselektivität für neue Liganden, neue Substratkombinationen und sogar für eine völlig neue Klasse von C–C‑Bindungsbildungen voraus, die nicht im Trainingssatz enthalten war. Die Analyse der wichtigsten Deskriptoren liefert zugleich Hinweise darauf, welcher Schritt im katalytischen Zyklus die Händigkeit für jede Reaktionsfamilie tatsächlich bestimmt.
Chemikern helfen, neue Reaktionen schneller zu starten
In einer abschließenden Demonstration verwenden die Autoren ihr Deskriptorschema zusammen mit einer bayesschen Optimierungsplattform, um eine nickelkatalysierte Kupplung von benzylischen Acetalen und Aryl‑iodiden zu entwerfen, die zuvor asymmetrisch nicht entwickelt worden war. Ausgehend von Literaturdaten zu anderen Reaktionen empfiehlt das Modell kleine Chargen vielversprechender Liganden zum Testen und findet in nur wenigen Dutzend Experimenten schnell die leistungsfähigste Klasse. Für einen Chemiker bedeutet das ein praktisches Werkzeug, um ein neues katalytisches Projekt schnell zu starten: Durch Eingabe einiger weniger Anfangsergebnisse kann das Modell vorschlagen, welche chiralen Liganden am ehesten hohe Enantioselektivität liefern. Insgesamt zeigt die Studie, dass wohlüberlegte, kostengünstige computergestützte Merkmale begrenzte vergangene Daten in breit anwendbare Orientierungshilfen für die Entwicklung der nächsten Generation selektiver Reaktionen verwandeln können.
Zitation: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Schlüsselwörter: asymmetrische Katalyse, Nickel-Cross-Coupling, Maschinelles Lernen in der Chemie, Reaktionsoptimierung, Prognose der Enantioselektivität