Clear Sky Science · de
Die entscheidende Rolle intrinsischer Defekte und Vielteilchenwechselwirkungen für die Stabilität von MnBi2Te4
Warum winzige Fehler in Kristallen für künftige Technologien wichtig sind
Viele der Quanten‑Technologien von morgen – etwa hocheffiziente Elektronik und neue Arten von Rechnern – beruhen auf exotischen Materialien, deren Oberflächen elektrischen Strom leiten, während das Innere isolierend bleibt. Eines der vielversprechendsten Beispiele ist MnBi2Te4, ein „topologischer Magnet“, der widerstandslose Randströme beherbergen könnte, nützlich für energiearme Bauelemente und Quantenberechnung. In realen Kristallen sitzen Atome jedoch häufig an falschen Stellen, und diese winzigen Fehler können die gewünschten Effekte stillschweigend zerstören. Die vorliegende Studie stellt eine einfache, aber zentrale Frage: Sind diese Fehlordnungen ein Herstellungsfehler oder werden sie bei den Temperaturen, bei denen das Material wächst, tatsächlich von der Natur bevorzugt?
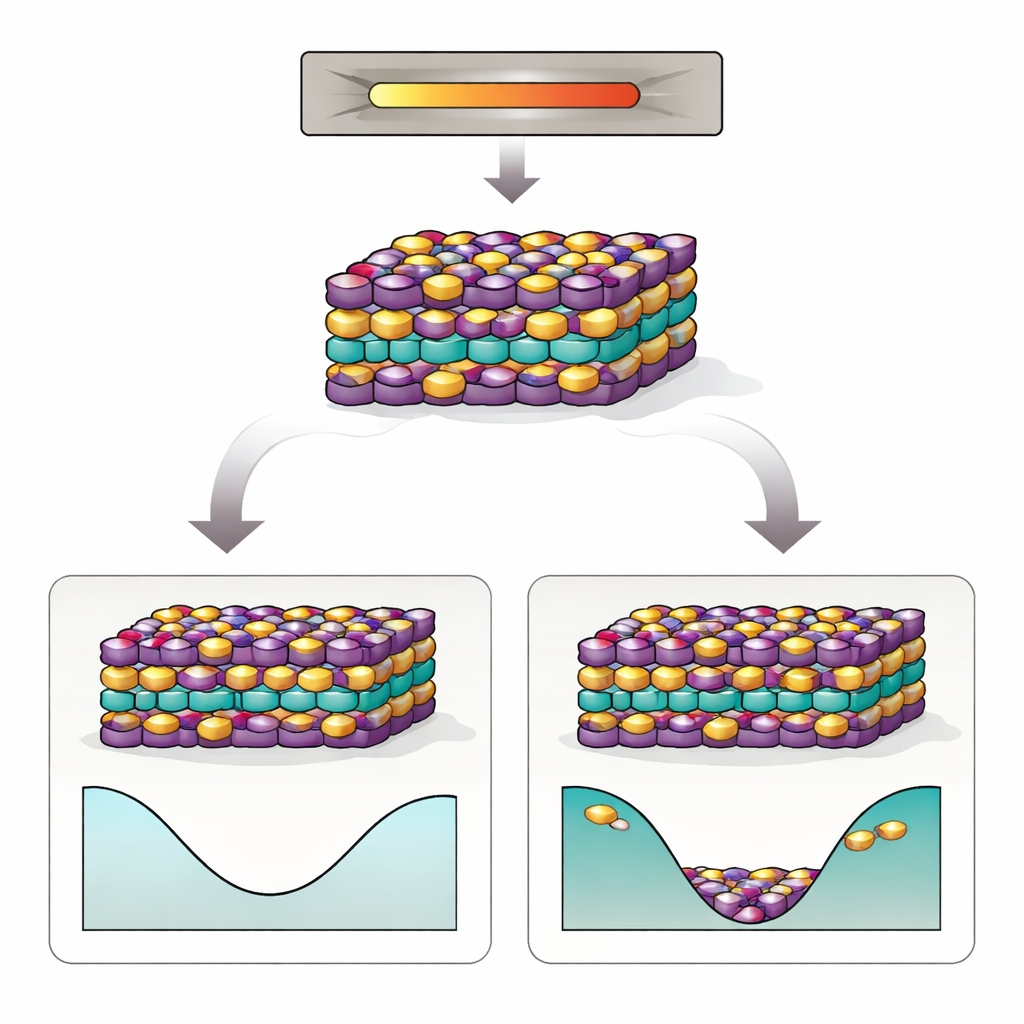
Ein vielversprechendes Material mit hartnäckigem Problem
MnBi2Te4 besteht aus gestapelten Atomlagen, ähnlich einem sorgfältig geschichteten Sandwich. Sein spezielles elektronisches Verhalten hängt von zwei Dingen ab: einer präzisen Anordnung von Mangan (Mn), Bismut (Bi) und Tellur (Te) sowie einem empfindlichen Muster magnetischer Ausrichtung zwischen den Schichten. In Experimenten findet man jedoch wiederholt, dass viele Mn‑ und Bi‑Atome ihre Plätze tauschen – sogenannte Antisiten‑Defekte. Diese Vertauschungen stören das magnetische Muster, treiben das Material vom idealen Isolatorzustand weg und erschweren das Beobachten der gewünschten Quantenphänomene. Schlimmer noch: Selbst wenn Kristalle mit großer Sorgfalt gezüchtet und annealiert werden, bleiben die Antisiten hartnäckig bestehen, was darauf hindeutet, dass etwas Tieferes als bloße Verarbeitungsfehler am Werk ist.
Warum frühere Rechnungen den Experimenten widersprachen
Standard‑Computersimulationen zeichneten ein rätselhaftes Bild. Bei absolutem Nullpunkt sagten gängige quantenmechanische Methoden voraus, dass die Erzeugung eines Mn–Bi‑Tauschs Energie kostet und daher selten sein sollte. Das steht im Widerspruch zu Experimenten, die hohe Defektraten in realen Proben zeigen, die bei rund 850 Kelvin (über 500 °C) hergestellt wurden. Die Autoren argumentieren, dass zwei entscheidende Aspekte in früheren Theorien fehlten. Erstens wurden Defekte meist einzeln behandelt, sodass ihre Wechselwirkung und mögliche Clusterbildung unberücksichtigt blieben. Zweitens wurden Rechnungen typischerweise bei Nulltemperatur durchgeführt und vernachlässigten, wie Wärme und Unordnung bevorzugte atomare Anordnungen verändern. In einem Material, das ohnehin nur marginal stabil ist, können selbst kleine Beiträge aus dem Vielteilchenverhalten der Elektronen und die schiere Anzahl möglicher Anordnungen das Gleichgewicht kippen.
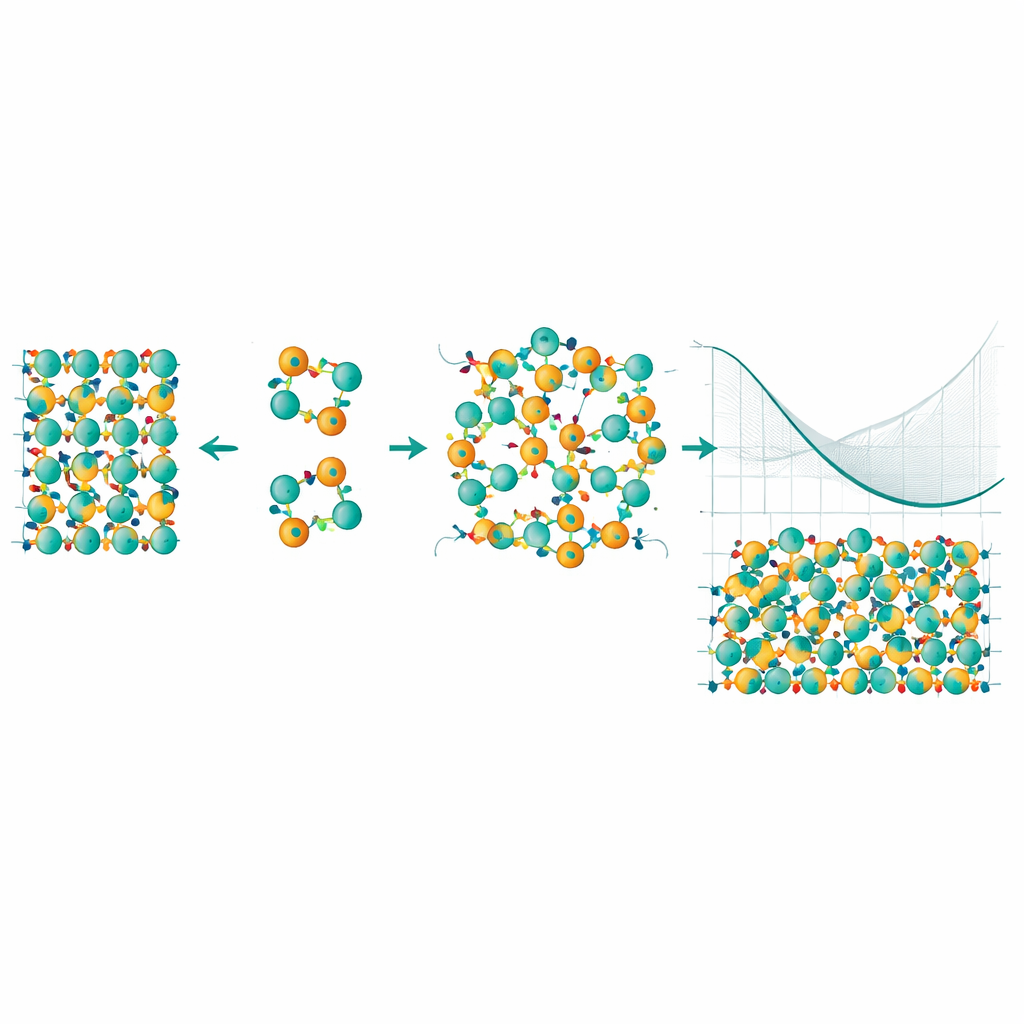
Jeden Tausch in einem virtuellen Kristall verfolgen
Um das anzugehen, bauten die Forscher ein statistisches Modell, das Millionen verschiedener Möglichkeiten untersucht, wie Mn‑ und Bi‑Atome sich umordnen könnten. Sie nutzten eine Technik namens Cluster‑Expansion, die die Energie des Kristalls in Beiträge von Einzelatomen, Paaren und kleinen Gruppen zerlegt, und kombinierten diese mit Monte‑Carlo‑Sampling, um zu sehen, welche Muster bei verschiedenen Temperaturen auftreten. Entscheidend war eine Korrektur der zugrunde liegenden Energien mit einer besonders genauen Methode, der Quanten‑Monte‑Carlo, die subtile Elektron‑Elektron‑Wechselwirkungen besser erfasst. Dieser hybride Ansatz ermöglichte es, nicht nur die Energiekosten eines einzelnen Tauschs zu berechnen, sondern auch wie sich diese Kosten ändern, wenn mehr Defekte auftreten und sich gegenseitig beeinflussen.
Wenn Unordnung die günstigere Option wird
Die Simulationen zeigen, dass Wechselwirkungen zwischen mehreren Antisiten‑Defekten und die „konfigurationsbedingte Entropie“ der Unordnung – im Wesentlichen die enorme Zahl von Möglichkeiten, die vertauschten Atome anzuordnen – das Verhalten des Materials bei Wachstums‑Temperaturen drastisch umformen. Obwohl ein einzelner Mn–Bi‑Tausch bei Nulltemperatur teuer ist, überwiegt bei höheren Temperaturen der Entropiegewinn diesen Energieaufwand. Die Autoren finden einen Ordnungs‑/Unordnungsübergang nahe der Synthesetemperatur: Oberhalb dieses Punkts werden vertauschte Mn‑ und Bi‑Atome thermodynamisch begünstigt, und die freie Energie eines defekten Kristalls fällt tatsächlich unter die eines perfekt geordneten. Anders gesagt: Die Natur bevorzugt einen Kristall mit einem erheblichen Anteil an Antisiten‑Defekten, und diese Defekte neigen dazu, in korrelierten Clustern zu entstehen statt rein zufällig.
Was das für die Herstellung besserer Quantenmaterialien bedeutet
Für Nicht‑Experten lautet die wichtigste Erkenntnis, dass die problematischen Defekte in MnBi2Te4 nicht einfach ein Herstellungsfehler sind; sie sind eine natürliche Folge der Thermodynamik des Materials bei den Temperaturen, bei denen es wächst. Die Studie zeigt, dass sich Theorie und Experiment endlich in Einklang bringen lassen, wenn Vielteilchenwechselwirkungen und die Statistik der Unordnung korrekt berücksichtigt werden: Antisiten‑Defekte entstehen spontan und in großer Zahl. Diese Einsicht erklärt, warum es so schwierig ist, wirklich defektfreie Kristalle herzustellen, und sie liefert einen Fahrplan zur Verbesserung anderer empfindlicher Quantenmaterialien. Jede Anstrengung, bessere Proben zu erzeugen – durch Änderung der Wachstumsbedingungen, Zusammensetzung oder Verarbeitungswege – muss berücksichtigen, dass Unordnung bei hohen Temperaturen keine Laune, sondern die energieärmste Wahl für den Kristall ist.
Zitation: Ghaffar, A., Saritas, K. & Reboredo, F.A. The critical role of intrinsic defects and many-body interactions on the stability of MnBi2Te4. npj Comput Mater 12, 119 (2026). https://doi.org/10.1038/s41524-026-02019-8
Schlüsselwörter: topologische Isolatoren, magnetische Materialien, Kristalldefekte, Quanten-Monte-Carlo, Materialthermodynamik