Clear Sky Science · de
Effiziente und genaue räumliche Mischung maschinell gelernter zwischenatomarer Potentiale für die Materialwissenschaft
Warum schnellere atomare Simulationen wichtig sind
Die Entwicklung besserer Werkstoffe für Technologien wie Kernfusion, Mikroelektronik und Strukturlegierungen beruht zunehmend auf Computersimulationen, die verfolgen, wie sich Atome bewegen und miteinander wechselwirken. Die genauesten Methoden übernehmen Konzepte aus der Quantenphysik, sind aber so rechenintensiv, dass nur moderate Systemgrößen und Zeiträume praktikabel sind. Dieser Artikel stellt ML‑MIX vor, eine Technik und eine Software, die es Forschern erlaubt, die nahezu quantenmechanische Genauigkeit genau dort beizubehalten, wo sie gebraucht wird, und gleichzeitig in anderen Bereichen einfachere, preiswertere Modelle einzusetzen. Das Ergebnis ist ein erheblicher Geschwindigkeitsgewinn – oft ein Faktor von 4 bis 10 – ohne Verlust der Zuverlässigkeit in den wichtigen physikalischen Vorhersagen.
Verschmelzen von detaillierten und einfachen Atomansichten
Im Kern steht eine einfache Idee: Nicht jedes Atom in einer Simulation braucht dieselbe Aufmerksamkeit. Regionen, in denen Bindungen sich dehnen, brechen oder umordnen – etwa Defekte, Oberflächen oder implantierte Partikel – profitieren von modernen, maschinell gelernten zwischenatomaren Potentialen, die quantenmechanische Genauigkeit nachahmen. Atome weit entfernt von diesen „Hotspots“ schwingen größtenteils um regelmäßige Positionen und können durch deutlich einfachere Modelle beschrieben werden. ML‑MIX bietet einen Weg, ein genaues, aber teures Modell mit einem sparsamen „günstigen“ Modell innerhalb desselben Simulationskastens zu kombinieren. Dazu definiert es eine Kernzone, die das teure Modell nutzt, einen umgebenden Puffer, in dem die Kräfte zwischen den Modellen sorgfältig gemischt werden, und eine äußere Bulk‑Zone, die nur die günstige Beschreibung verwendet. 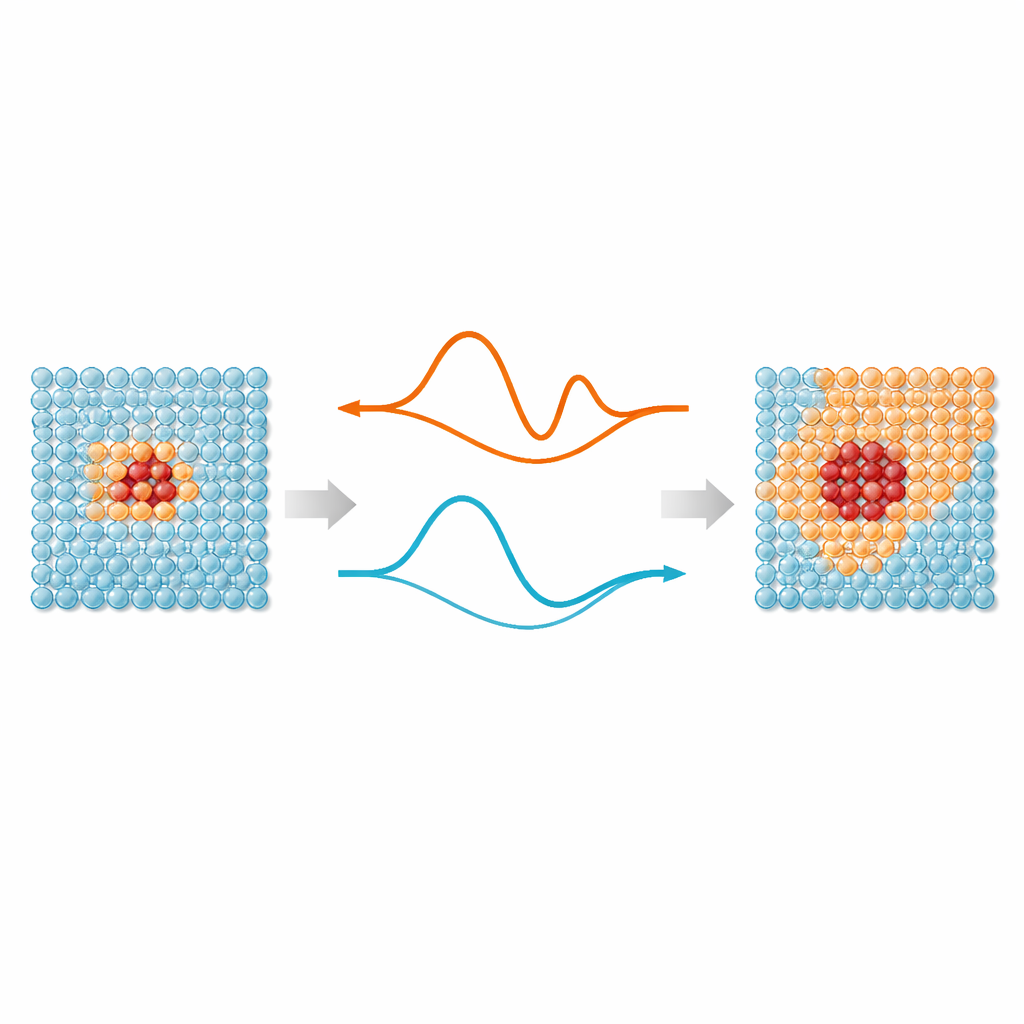
Ein günstiges Modell lehren, das genaue zu imitieren
Eine zentrale Herausforderung ist sicherzustellen, dass sich das günstige Modell dort, wo die Modelle aufeinandertreffen, wie das genaue verhält. Anstatt das günstige Modell direkt an einen riesigen und vielfältigen quantenmechanischen Datensatz anzupassen, erzeugen die Autoren fokussierte „synthetische“ Daten, indem sie das genaue Modell unter den spezifischen Bedingungen laufen lassen, die für die Bulk‑Region relevant sind: hochtemperaturbedingte Schwingungen und sanft gedehnte Kristalle. Anschließend passen sie das günstige Modell so an, dass es diese Daten reproduziert, während sie strikte Nebenbedingungen für grundlegende Materialeigenschaften wie Elastizitätskonstanten und Gitterabstände erzwingen. Diese eingeschränkte Anpassung stellt sicher, dass langreichweitige Spannungen und Dehnungen glatt über die Grenze zwischen den beiden Modellen hinweg übereinstimmen und künstliche Kräfte vermieden werden, die die Dynamik in der Nähe der Schnittstelle verfälschen könnten.
Die Methode auf die Probe stellen
Um zu prüfen, dass ML‑MIX wirklich funktioniert, führen die Autoren eine Reihe von Tests an Silizium-, Eisen‑ und Wolfram‑Systemen durch. Als einfaches Beispiel berechnen sie die Energiebarriere für eine Vakanz – eine leere Gitterstelle – in Silizium, um von einer Position zur anderen zu wandern. Die gemischte Simulation reproduziert das Ergebnis einer vollständig teuren Berechnung bis auf ein Tausendstel eines Elektronvolt, läuft dabei aber etwa fünfmal schneller. In einer dynamischeren Einstellung dehnen sie eine einzelne Siliziumbindung in einem heißen Kristall und messen die mittlere auf sie wirkende Kraft. Eine Simulation, die nur das günstige Modell verwendet, kommt überraschend nahe, doch sobald um die gedehnte Bindung ein kleiner teurer Kern hinzukommt, wird die Übereinstimmung statistisch nicht mehr von der vollständig genauen Referenz zu unterscheiden, mit Beschleunigungen von bis zu einem Faktor von etwa 13 in seriellen Läufen.
Defekte und Partikel in Bewegung verfolgen
Realistischere Tests untersuchen, wie sich Defekte in Metallen bewegen. Das Team simuliert die Diffusion eines Selbst‑Interstitial‑Defekts in Eisen und von Heliumatomen in Wolfram. In jedem Fall ist das teure Modell auf eine kleine bewegliche Region um den Defekt beschränkt, während der Rest des Kristalls vom günstigen Potential behandelt wird. Die resultierenden Diffusionskoeffizienten stimmen innerhalb statistischer Fehler mit denen vollständig genauer Simulationen überein, selbst wenn eine ausschließlich günstige Simulation versagt hätte. Die Autoren wenden die Methode dann auf größere, wissenschaftlich relevante Probleme in Wolfram an, einem führenden Kandidatenmaterial für Fusionsreaktoren. Sie modellieren die Bewegung von Schraubversetzungen – linienartige Defekte, die die plastische Verformung steuern – sowie die Implantation von Heliumatomen in eine heiße Wolframoberfläche. In beiden Fällen reproduziert ML‑MIX die Ergebnisse rein teurer Rechnungen und reduziert die Rechenkosten um Faktoren von etwa vier bis elf. 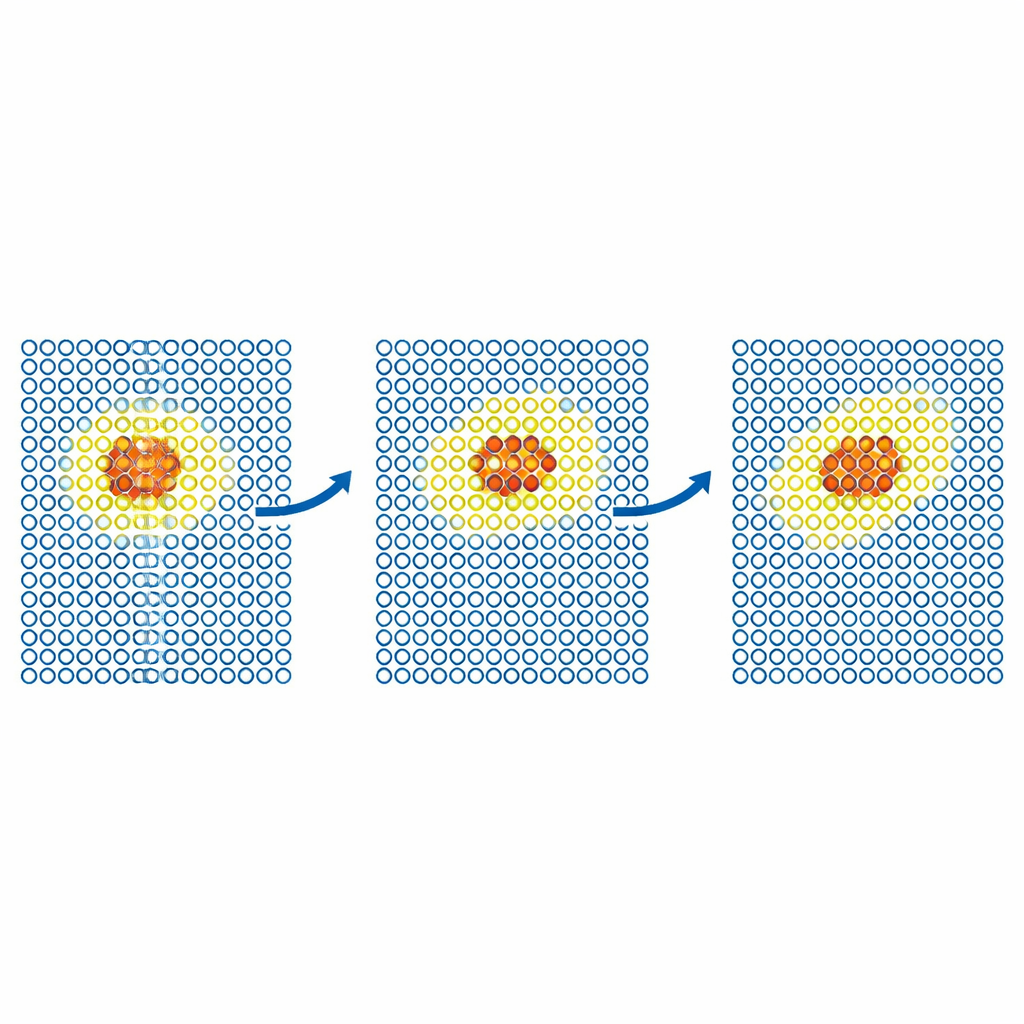
Experimente abgleichen und ein Blick nach vorn
Die Studie zur Heliumimplantation zeigt die Stärke dieses Ansatzes besonders deutlich. Indem ein hochmodernes maschinell gelerntes Modell für Helium‑Wolfram‑Wechselwirkungen mit einem schnelleren Potential für reines Wolfram gemischt wird, können die Autoren viel mehr Einschlagsereignisse und größere Proben simulieren, als sonst möglich wäre – und das alles auf Grafikkarten. Der prognostizierte Anteil der Heliumatome, die von der Oberfläche abprallen statt sich im Metall zu implantieren, stimmt mit experimentellen Messungen bis zu Einfallsenergien von etwa 80 Elektronvolt überein, etwas, das frühere Simulationen schwer erreichten. Obwohl das Mischschema nicht strikt Energie konserviert und sanfte Thermostate erfordert, ist die resultierende Drift klein und handhabbar. Insgesamt zeigt ML‑MIX, dass eine sorgfältige Kombination detaillierter und vereinfachter atomarer Modelle langjährige Schranken zwischen Genauigkeit und Umfang durchbrechen kann und den Weg für routinemäßige, hochaufgelöste Simulationen komplexer Materialien in realistischen Umgebungen öffnet.
Zitation: Birks, F., Nutter, M., Swinburne, T.D. et al. Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science. npj Comput Mater 12, 110 (2026). https://doi.org/10.1038/s41524-026-01982-6
Schlüsselwörter: maschinell gelernte zwischenatomare Potentiale, multiskalige Materialsimulation, Wolfram-Helium-Implantation, Defekte und Versetzungen, Beschleunigung molekularer Dynamik