Clear Sky Science · de
Graph-atomare Cluster-Erweiterung für fundamentale Machine-Learning-Zwischenatompotenziale
Computern beibringen, die Atome zu „fühlen“
Die Entwicklung neuer Materialien für Batterien, Flugzeuge oder Fusionsreaktoren läuft oft auf eine einfache Frage hinaus: Wie ziehen und drücken sich Atome gegenseitig? Diese Kräfte exakt zu berechnen ist so aufwendig, dass es für ein einziges Material Tage auf einem Supercomputer dauern kann. Dieses Paper stellt eine neue Familie von Machine-Learning-Modellen vor, genannt GRACE, die wie ein universeller „Rechner“ für atomare Kräfte über einen Großteil des Periodensystems funktionieren sollen. Ziel ist es, genaue Simulationen komplexer Materialien zur Routine statt zur Heldentat zu machen.
Ein einziges Modell für viele Materialien
Die meisten bestehenden Machine-Learning-Kraftfelder sind Spezialistenwerkzeuge: Sie arbeiten sehr gut für einige wenige Elemente oder Verbindungen, müssen aber beim Hinzufügen neuer Elemente von Grund auf neu aufgebaut werden. GRACE geht einen anderen Weg. Es ist von vornherein als ein grundlegendes Modell konzipiert, das 89 chemische Elemente und eine enorme Vielfalt atomarer Anordnungen mit einem gemeinsamen Regelwerk behandeln kann. Dafür bauen die Autorinnen und Autoren auf einem mathematischen Rahmen auf, der als atomare Cluster-Erweiterung bekannt ist, und erweitern ihn auf graphähnliche Strukturen. So kann das Modell sowohl lokale Nachbarschaften von Atomen als auch ausgedehntere Muster einheitlich beschreiben. Anstatt jede mögliche Wechselwirkung hart zu kodieren, lernt GRACE kompakte „Embedding“-Darstellungen, die Ähnlichkeiten zwischen Elementen erfassen, sodass Wissen über ein Material bei der Beschreibung eines anderen helfen kann.
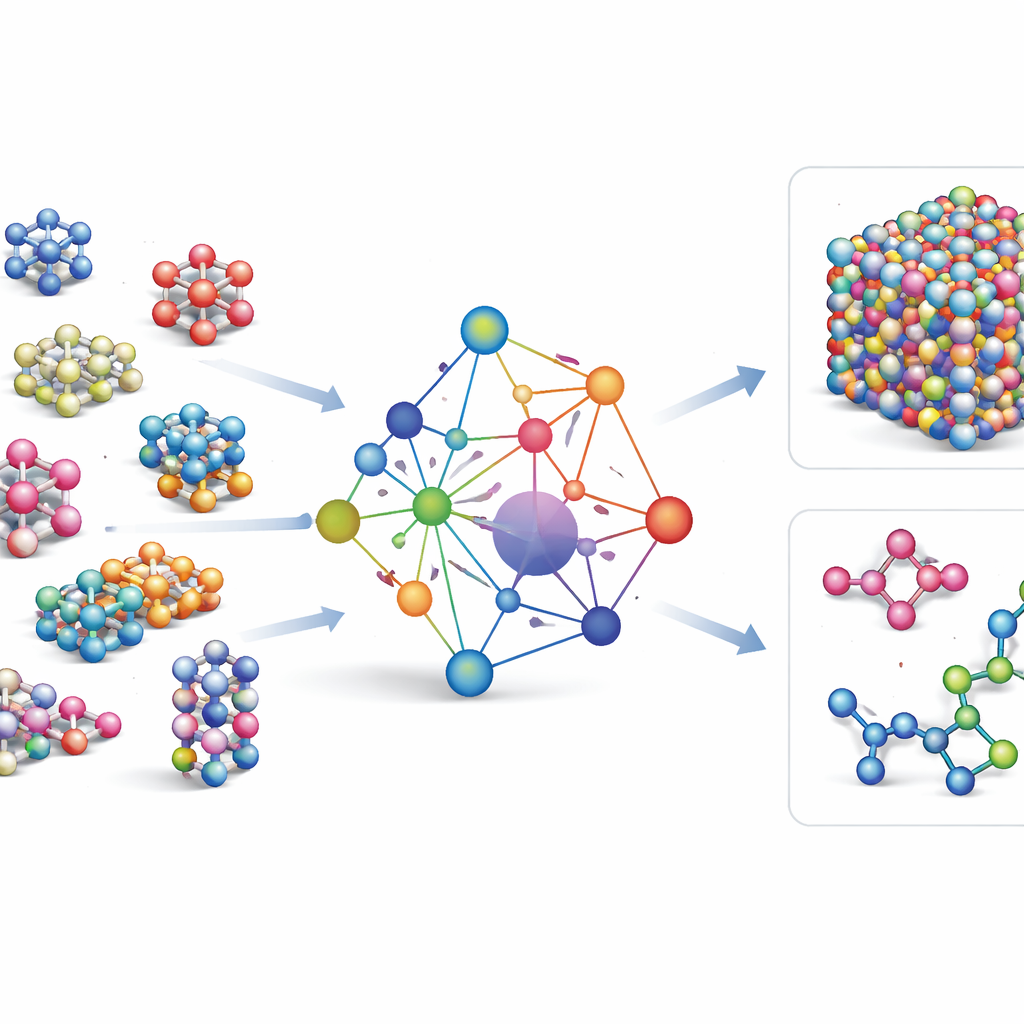
Training im Meer atomarer Daten
Um GRACE beizubringen, wie sich Atome verhalten, stellten die Autorinnen und Autoren einige der größten öffentlichen Datenbanken quantenmechanischer Rechnungen zusammen. Der Kern ist die OMat24-Sammlung, die rund 110 Millionen Simulationen anorganischer Materialien enthält, ergänzt durch zwei weitere Datensätze, die verfolgen, wie sich Strukturen entspannen und entwickeln. Zusammen decken diese Datensätze nahezu-gleichgewichtsnahen Kristalle, gestauchte und verzerrte Strukturen, hochtemperierte Flüssigkeiten und mehr ab, über denselben breiten Elementsatz hinweg. GRACE-Modelle gibt es in mehreren Größen, von einfachen einlagigen Versionen, die nur lokale atomare Umgebungen betrachten, bis hin zu tieferen zweilagigen Varianten, die effektiv „Nachrichten“ zwischen benachbarten Bereichen weitergeben. Das anfängliche Training zielt auf ein ausgewogenes Verhältnis von Energien, Kräften und inneren Spannungen; ein weiteres Feintuning passt die Modelle an, sodass sie mit weit verbreiteten Referenzdatenbanken der Materialwissenschaft kompatibel sind.
Das Modell auf die Probe stellen
Ein universelles Modell ist nur nützlich, wenn es zuverlässig über viele Aufgaben hinweg Leistung zeigt. Die Autorinnen und Autoren unterzogen GRACE daher einer anspruchsvollen Testreihe, die widerspiegelt, wie Wissenschaftlerinnen und Wissenschaftler atomistische Simulationen tatsächlich verwenden. In einem Community-Benchmark zur Entdeckung stabiler Kristallstrukturen befindet sich GRACE konsistent auf der Pareto-Front: Bei gegebener Genauigkeit ist es schneller als konkurrierende Modelle, und bei gegebener Geschwindigkeit ist es genauer. Ähnliche Vorteile zeigen sich bei der Vorhersage der thermischen Leitfähigkeit, einer Eigenschaft, die sehr empfindlich auf kleine Änderungen in der atomaren Bewegung reagiert. GRACE schneidet auch gut bei elastischen Eigenschaften, Oberflächenenergien, Korngrenzenenergien und Bildungsenergien Punktdefekte in vielen reinen Metallen ab — alles Untersuchungen dafür, wie Materialien auf Dehnung, Zerschneiden oder lokale Schäden reagieren. Ein längerer Molekulardynamik-Lauf eines heißen geschmolzenen Salzes zeigt, dass das Modell für Nanosekunden numerisch stabil bleibt, während es detaillierte Strukturmuster und atomare Diffusionsraten reproduziert.
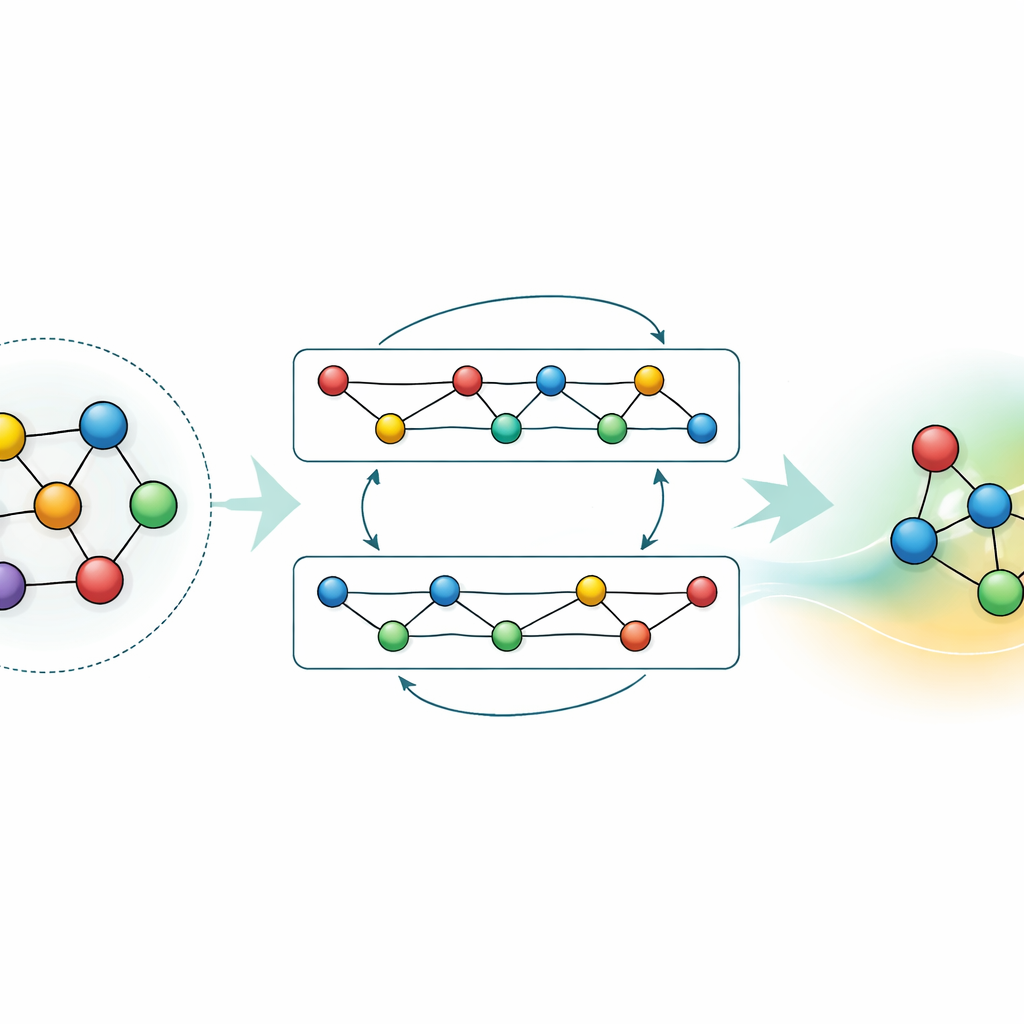
Wissen anpassen und komprimieren
Während ein Generalmodell mächtig ist, benötigen viele Anwendungen entweder höhere Genauigkeit für ein bestimmtes Material oder schnellere Berechnungen auf bescheidener Hardware. Die Autorinnen und Autoren demonstrieren zwei Strategien, um dies zu erreichen, ohne das bereits von GRACE Gelernte wegzuwerfen. Erstens feinjustieren sie das fundamentale Modell auf fokussierten Datensätzen, etwa Aluminium-Lithium-Legierungen oder detaillierten Wasserstoff-Verbrennungswegen. Für die Legierungen verschärfen selbst moderate zusätzliche Daten die Vorhersagen deutlich und übertreffen Modelle, die mit denselben Informationen von Grund auf neu trainiert wurden. Bei der Verbrennung würde ein naives Fine-Tuning das Modell normalerweise dazu bringen, sein Wissen über andere Materialien zu „vergessen“; durch das sorgfältige Einfrieren von Teilen des Netzwerks und das Aktualisieren nur ausgewählter Parameter begrenzen die Autorinnen und Autoren dieses katastrophale Vergessen, während sie dennoch Genauigkeit für die neue Chemie gewinnen. Zweitens zeigen sie, wie das große Modell in einen viel einfacheren „Studenten“ destilliert werden kann, der den Lehrer in Schlüsselbereichen nachahmt. Diese destillierte Version läuft auf einer CPU ungefähr siebzigmal schneller und bewahrt dennoch den Großteil der Genauigkeit, besonders wenn sie auf einer Mischung aus komplexen Legierungen und einfacheren Referenzstrukturen trainiert wird, die vom ursprünglichen GRACE gelabelt wurden.
Was das für die zukünftige Materialentwicklung bedeutet
Die Arbeit positioniert GRACE als flexible Grundlage für die nächste Generation atomistischer Modellierung. Anstatt für jedes Material oder jeden gewünschten Eigenschaftenbereich ein neues Potential zu entwickeln, können Forschende von einem universellen GRACE-Modell ausgehen und es dann feinjustieren oder destillieren, um ihren Bedürfnissen gerecht zu werden — was enorme Einsparungen an Rechenzeit und Expertenaufwand bedeutet. Die Benchmarks zeigen, dass dieser Ansatz bestehende Werkzeuge nicht nur erreicht, sondern sie oft in Geschwindigkeit und Zuverlässigkeit übertrifft, insbesondere bei anspruchsvollen Eigenschaften wie dem thermischen Transport. Für Nicht-Spezialisten lautet die Kernbotschaft: Ein einziges, gut gestaltetes Machine-Learning-Modell kann jetzt als breit vertrauenswürdige „Maschine" für virtuelle Experimente über einen Großteil des Periodensystems dienen und die Suche nach besseren Batterien, Katalysatoren, Strukturlegierungen und Energiematerialien beschleunigen.
Zitation: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
Schlüsselwörter: machine learning zwischenatomare Potentiale, Materialmodellierung, Atomsimulationen, fundamentale Modelle, graph-atomare Cluster-Erweiterung