Clear Sky Science · de
Selbstoptimierender, durch maschinelles Lernen unterstützter automatisierter Workflow für hocheffizientes Design komplexer Systemmaterialien
Intelligentere Suche nach neuen Materialien
Neue Materialien zu entwerfen ist wie die Suche nach einer Nadel in einem nahezu unendlichen Heuhaufen. Von besseren Batterien und schnelleren Rechnern bis hin zu effizienteren Lasern und potenziellen Supraleitern bei Raumtemperatur: Viele Zukunftstechnologien hängen davon ab, genau die richtigen atomaren Anordnungen zu finden. Dieser Beitrag stellt eine Methode vor, mit der künstliche Intelligenz den Großteil dieser Suche automatisch übernehmen kann und so Zeit und Kosten für die Entdeckung vielversprechender neuer Verbindungen drastisch reduziert.
Warum das Materialrätsel so schwierig ist
Die Eigenschaften eines Festkörpers — wie gut er Strom leitet, wie stabil er ist, wie er auf Licht reagiert — werden durch die Anordnung seiner Atome in dreidimensionalen Mustern bestimmt, den Kristallstrukturen. Theoretisch lassen sich mittels Quantenmechanik berechnen, welche Anordnungen stabil sind und welche Eigenschaften sie haben. In der Praxis sind diese Quantenberechnungen so aufwändig, dass nur ein winziger Bruchteil aller möglichen Materialien geprüft werden kann. Die Herausforderung wächst rapide, wenn mehr als zwei chemische Elemente beteiligt sind, denn dann explodiert die Zahl der Kombinationen und atomaren Anordnungen, wodurch eine blinde Suche unmöglich wird.
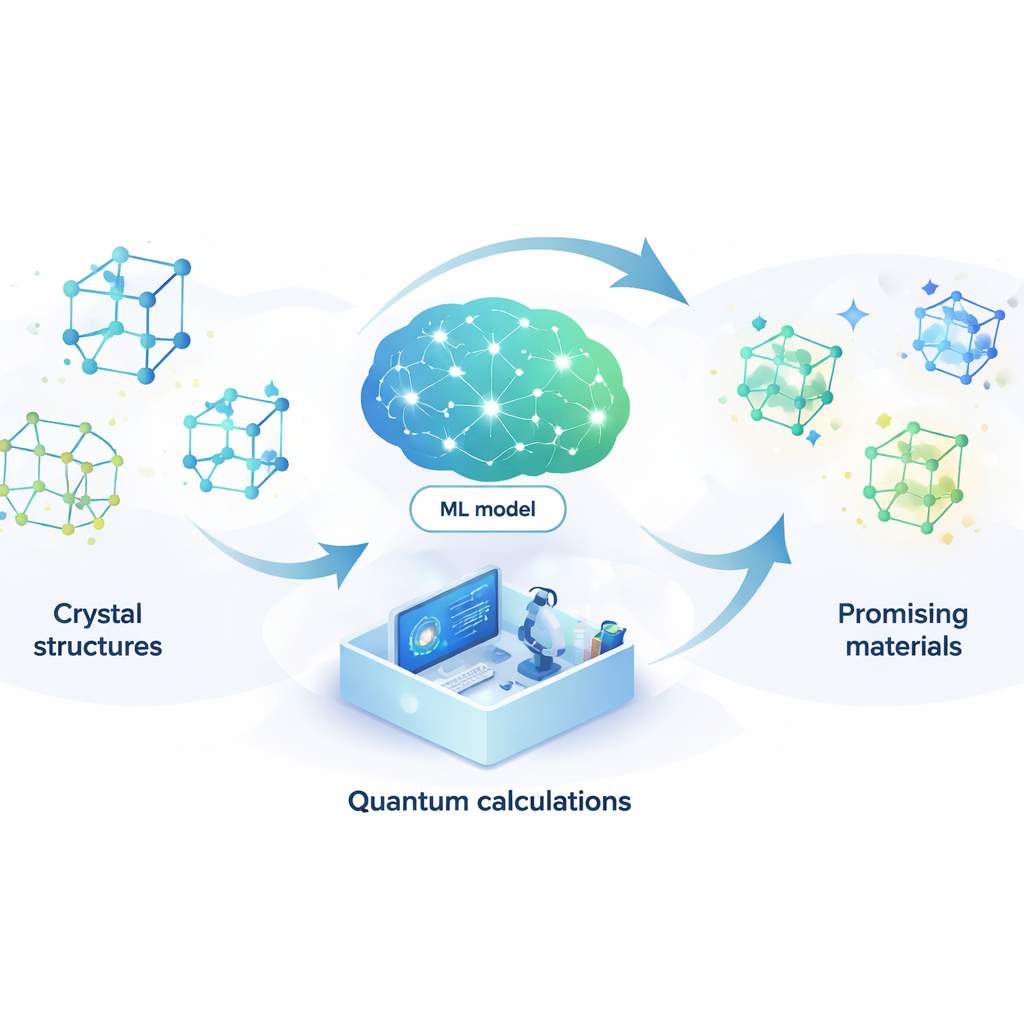
Ein Lernmodell anstelle der Quantenphysik
Um dieses Problem anzugehen, bauen die Autoren ein Modell des maschinellen Lernens, das die Ergebnisse teurer Quantenberechnungen zu einem Bruchteil der Kosten nachahmen kann. Ihr Modell, ein attention-gekoppeltes neuronales Netzwerk (ACNN), lernt, wie die Energie eines Materials von den Positionen und Typen seiner Atome abhängt. Einmal trainiert, kann es sehr schnell einschätzen, ob eine vorgeschlagene Kristallstruktur wahrscheinlich stabil ist und welche Kräfte auf die einzelnen Atome wirken. Entscheidend ist, dass das Modell so gestaltet ist, dass es grundlegende physikalische Forderungen respektiert — etwa dass eine Verschiebung oder Rotation des gesamten Kristalls seine Gesamtenergie nicht verändern darf.
Eine sich selbst verbessernde Materials-Entdeckungsschleife
Anstatt das Modell einmal zu trainieren und zu hoffen, dass es überall funktioniert, binden die Autoren es in eine selbstoptimierende Schleife ein. Der Prozess beginnt mit einer kleinen Menge zufälliger Kristallstrukturen, die mit vollständigen quantenmechanischen Berechnungen bewertet und zum Training eines initialen ACNN genutzt werden. Dieses Modell wird dann verwendet, um Millionen von Strukturvorschlägen zu entspannen und so schnell lokale Energie-Minima zu finden — Kandidaten für stabile oder nahezu stabile Phasen. Der Workflow markiert automatisch zwei besonders wertvolle Strukturtypen: solche, die sehr stabil aussehen, und solche, die unphysikalisch oder verdächtig erscheinen. Nur diese ausgewählten Fälle werden zurück an den teuren Quantenlöser geschickt, und die neuen Ergebnisse fließen ins Modell für ein erneutes Training ein. Über viele Runden wird das Modell in den für die Suche relevanten Bereichen des Strukturraums immer genauer.
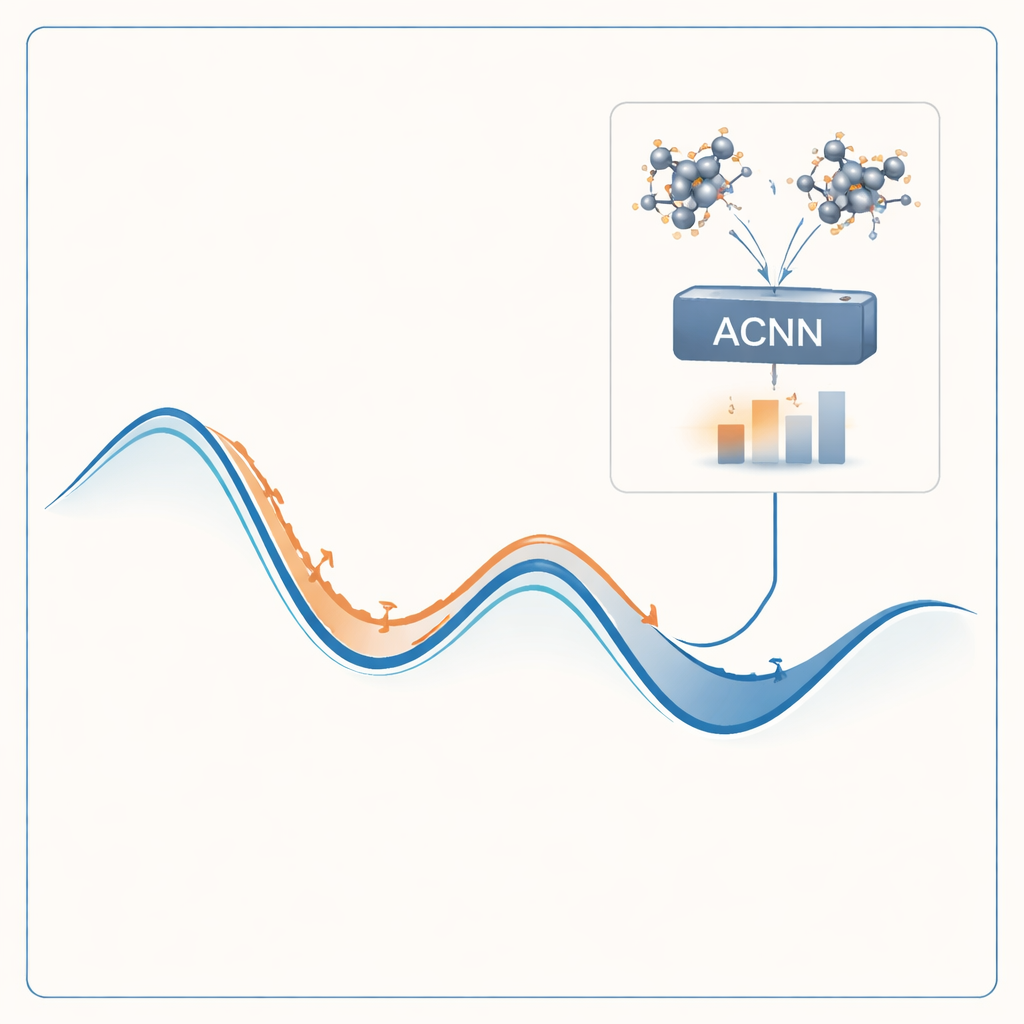
Prüfung der Methode
Das Team demonstriert seinen Ansatz an zwei anspruchsvollen Systemen. Das erste ist ein Hochdruckgemisch aus Magnesium, Calcium und Wasserstoff — eine Verbindungsfamilie von großem Interesse für hochtemperaturige Supraleitung. Durch die Untersuchung von fast sechs Millionen Strukturvorschlägen entdeckt ihr Workflow eine neue stabile Phase, MgCa₃H₂₃, und mehrere eng verwandte wasserstoffreiche „Käfig“-Strukturen. Rechnungen deuten darauf hin, dass einige davon unter extremem Druck bei Temperaturen über dem Siedepunkt von flüssigem Stickstoff supraleitend sein könnten. Der zweite Test richtet sich auf ein Vier-Elemente-System mit Beryllium, Phosphor, Stickstoff und Sauerstoff, ausgewählt wegen seines Potenzials, Kristalle zu beherbergen, die Laserlicht effizient in tiefes Ultraviolett umwandeln. Hier entspannt die Methode mehr als neun Millionen Strukturen und identifiziert drei thermodynamisch stabile Phasen mit sehr großen Bandlücken und vielversprechenden optischen Eigenschaften.
Von roher Gewalt zu gesteuerter Entdeckung
In beiden Beispielen erzielt der automatisierte Workflow eine Beschleunigung um etwa das Zehntausendfache im Vergleich zu rein quantenmechanischen Berechnungen, während er dennoch zuverlässig Strukturen identifiziert, die eine nähere Untersuchung verdienen. Für Nichtfachleute lautet die zentrale Botschaft: Ein großer Teil der Materialentdeckung lässt sich nun von einem lernenden System übernehmen, das seine Unsicherheiten erkennt und nur gezielt hochpräzise Berechnungen anfordert, wenn sie nötig sind. Diese Art selbstkorrigierender, KI-gestützter Suche öffnet die Tür zur Erforschung deutlich komplexerer Elementmischungen als bisher möglich und erhöht die Chancen, neue Supraleiter, optische Kristalle und andere funktionale Materialien zu finden, die den Kern zukünftiger Technologien bilden.
Zitation: Li, J., Feng, J., Luo, J. et al. Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design. npj Comput Mater 12, 101 (2026). https://doi.org/10.1038/s41524-026-01971-9
Schlüsselwörter: Materialentdeckung, maschinelle Lernpotenziale, Vorhersage von Kristallstrukturen, supraleitende Hydride, nichtlineare optische Kristalle