Clear Sky Science · de
JAK/STAT1-Interferon-ISGylierungs-Netzwerke bei der Resistenz von Brustkrebs gegen Inhibitoren von FOXM1 und CDK4/6
Warum das für die Behandlung von Brustkrebs wichtig ist
Viele Frauen mit östrogenrezeptorpositivem (ER+) Brustkrebs erhalten heute gezielte Medikamente, die das Tumorwachstum verlangsamen, indem sie den Zellzyklus stoppen. Doch fast unvermeidlich lernen Tumoren, diese Therapien zu umgehen, und wachsen wieder. Diese Studie stellt eine drängende Frage: Wenn ER+ Brustkrebse gegenüber zwei wichtigen Wirkstoffklassen — FOXM1-Inhibitoren und CDK4/6-Inhibitoren — resistent werden, welche innerzellulären Veränderungen erlauben ihnen die Flucht, und können diese Veränderungen neue Angriffspunkte liefern?
Eine gemeinsame Überlebensverdrahtung in resistenten Tumoren
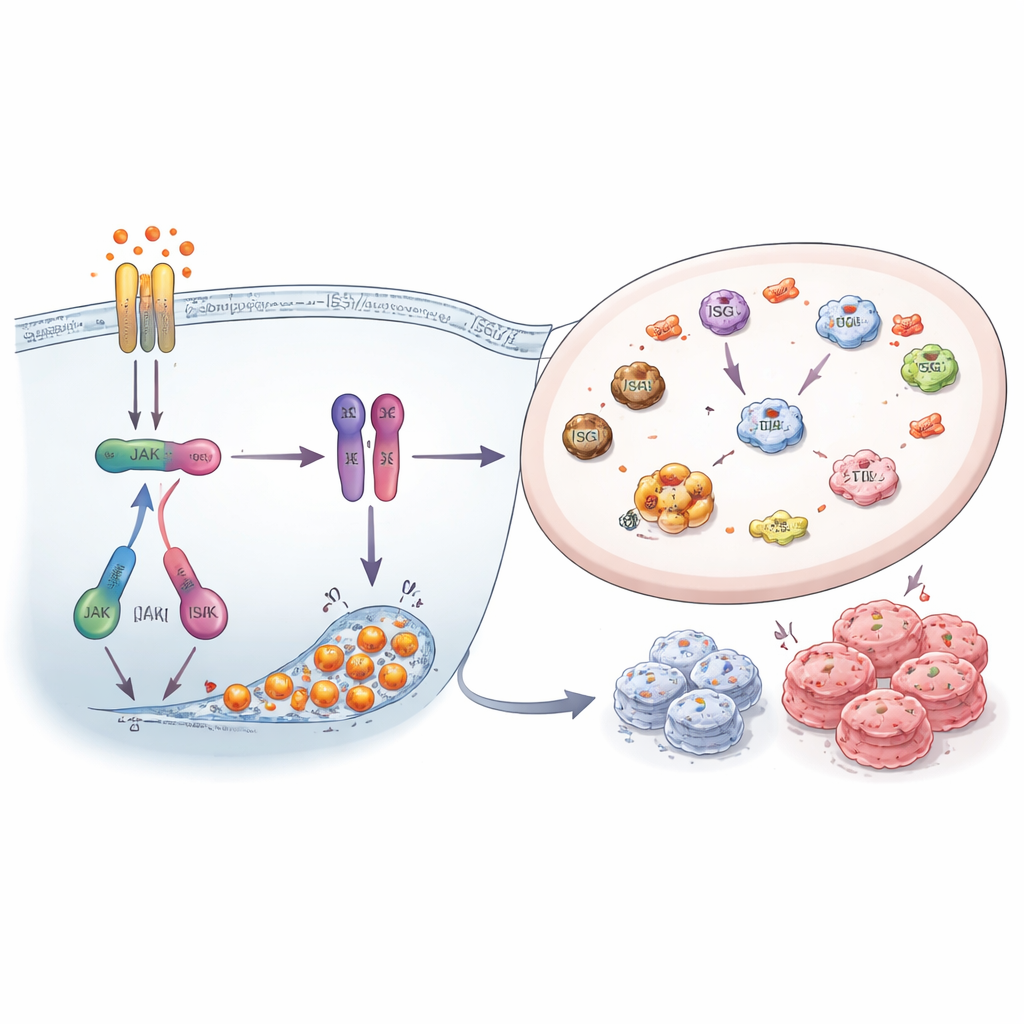
Die Forscher konzentrierten sich auf ER+ Brustkrebszellen, die im Labor so lange kultiviert wurden, bis sie nicht mehr auf entweder FOXM1-blockierende oder CDK4/6-blockierende Medikamente wie Palbociclib und Abemaciclib reagierten. Sie stellten fest, dass diese Zellen, obwohl sie gegen unterschiedliche Wirkstoffe resistent gemacht worden waren, ein ähnliches internes Alarmsystem aktivierten, das um Interferonsignale und das Protein STAT1 aufgebaut ist. Dieser Alarm führt zur Produktion vieler „Interferon-stimulierten Gene“ und eines kleinen Proteins namens ISG15, das wie eine molekulare Markierung an andere Proteine angehängt werden kann. Resistente Zellen wiesen deutlich höhere Mengen an STAT1, aktiviertem STAT1, freiem ISG15 und ISG15-markierten Proteinen auf als ihre nicht-resistenten Gegenstücke, was darauf hindeutet, dass dieses Netzwerk eine gemeinsame Basis der Arzneimittelresistenz bildet.
Ein schützender Mantel aus molekularen Markierungen
Bei genauerer Betrachtung sah das Team, dass die resistenten Zellen nicht nur mehr ISG15 produzierten, sondern auch Enzyme hochregulierten, die ISG15 an andere Proteine anfügen — ein Prozess, der als ISGylierung bekannt ist. Diese Enzyme — HERC5, HERC6 und UBE2L6 — waren stark erhöht, insbesondere in Zellen, die gegen FOXM1-Inhibitoren resistent waren. Viele zelluläre Proteine, einschließlich STAT1 selbst, trugen in resistenten Zellen ISG15-Markierungen, oft in höheren Mengen als in den ursprünglich medikamentensensitiven Zellen. Da das Anfügen dieser Markierungen die Stabilität und Funktion von Proteinen verändern kann, scheint die Anhäufung von ISGylierten Proteinen Teil des Mechanismus zu sein, mit dem Krebszellen sich gegenüber Therapien wappnen.
Das Ausschalten des Alarms schwächt resistente Zellen
Die Untersuchenden fragten dann, ob das Herunterregulieren dieses Alarmnetzwerks resistente Zellen verwundbarer macht. Sie verwendeten Wirkstoffe, die JAK-Kinasen blockieren — Schlüssel-Schalter upstream von STAT1 — sowie kleine interferierende RNAs, um ISG15, HERC5 und HERC6 zu reduzieren. Die Blockade der JAK1/2-Signalgebung senkte die STAT1-Aktivität deutlich, reduzierte ISG15-Level und ISGylierung und verringerte die Koloniebildung resistenter Zellen, vor allem jener, die gegen FOXM1-Inhibitoren resistent waren. Ebenso führte die direkte Stilllegung von ISG15 und seinen Anfügeenzymen zu einer Abnahme des ISGylierungsmusters und beeinträchtigte das Überleben der Zellen. Diese Experimente zeigen, dass das Interferon–STAT1–ISG15-System nicht nur ein Nebenphänomen ist, sondern aktiv das Wachstum und die Persistenz medikamentenresistenter Krebszellen unterstützt.
Neue Hoffnungen für sequenzielle Behandlungsstrategien
Eines der ermutigendsten Ergebnisse ist, dass die resistenten Zellen nicht in einem einzigen unbesiegbaren Zustand feststeckten. Brustkrebszellen, die gegen CDK4/6-Inhibitoren resistent geworden waren, reagierten noch auf FOXM1-Inhibitoren, und Zellen, die gegen FOXM1-Inhibitoren resistent waren, ließen sich weiterhin durch Palbociclib oder Abemaciclib verlangsamen. Sowohl in zweidimensionalen Zellkulturen als auch in dreidimensionalen Matrigel-Kulturen, die Tumoren besser nachahmen, reduzierte der Wechsel der Wirkstoffklasse das Zellwachstum deutlich und senkte die Expression von Genen, die Kopieren der DNA und Zellteilung antreiben. Gleichzeitig zeigten Patientendaten, dass hohe Level von ISG15 und seinen zugehörigen Enzymen in ER+/HER2− Tumoren mit schlechterem Überleben verbunden sind, was die klinische Relevanz dieser Resistenzverdrahtung unterstreicht.
Was das für Patientinnen und zukünftige Therapien bedeutet
Für eine nichtwissenschaftliche Leserin ergibt sich das Bild, dass resistente ER+ Brustkrebse sich um einen gemeinsamen Stressantwortkreis herum neu verdrahten und Interferonsignale sowie ISG15-Markierungen als eine Art schützende Rüstung nutzen. Die gute Nachricht ist, dass diese Rüstung neue Schwachstellen offenlegt. Wirkstoffe, die den JAK–STAT1–ISG15-Weg blockieren, sowie Therapiepläne, die FOXM1-Inhibitoren nach CDK4/6-Medikamenten (oder umgekehrt) sequenziell einsetzen, könnten helfen, der Resistenz zuvorzukommen, statt von ihr überwunden zu werden. Obwohl diese Erkenntnisse noch in klinischen Studien geprüft werden müssen, liefern sie eine klarere Roadmap, wie ein häufiges Therapietotpunkt in eine neue Chance zur Krankheitskontrolle verwandelt werden kann.
Zitation: Ziegler, Y., Kumar, S., Saeh, C.M. et al. JAK/STAT1-interferon-ISGylation networks in breast cancer resistance to inhibitors of FOXM1 and CDK4/6. npj Breast Cancer 12, 44 (2026). https://doi.org/10.1038/s41523-026-00911-6
Schlüsselwörter: ER-positiver Brustkrebs, Arzneimittelresistenz, CDK4/6-Inhibitoren, FOXM1-Inhibitoren, Interferon-Signalgebung