Clear Sky Science · de
Dateneffiziente Erlernung grobkörniger Molekulardynamik aus Kräften und Rauschen
Warum das Verkleinern von Molekülen wichtig ist
Die Simulation der unablässigen Bewegung jedes Atoms in einem Protein und seinem umgebenden Wasser gehört zu unseren besten Werkzeugen, um zu verstehen, wie Leben auf molekularer Ebene funktioniert. Doch diese all‑atom‑Simulationen sind so rechenintensiv, dass das Verfolgen eines Proteins beim Falten, Entfalten oder bei Wechselwirkungen mit Partnern über biologisch relevante Zeiträume Monate auf einem Supercomputer dauern kann. Dieser Artikel stellt eine neue Methode vor, um schnelle, vereinfachte Modelle von Proteinen zu bauen, die sich dennoch wie ihre vollständigen atomaren Gegenstücke verhalten und dabei deutlich weniger Trainingsdaten und Rechenleistung benötigen als bisher.
Vom einzelnen Atom zu einem einfacheren Bild
Die traditionelle Molekulardynamik verfolgt jedes Atom und berechnet die Kräfte zwischen ihnen in jedem winzigen Zeitschritt. Um die Rechenzeit zu verkürzen, verwenden Wissenschaftler häufig grobkörnige Modelle, bei denen viele Atome zu einer kleineren Anzahl von „Kügelchen“ zusammengefasst werden. Diese reduzierten Modelle laufen deutlich schneller, hatten aber historisch Schwierigkeiten, die Genauigkeit vollständiger atomistischer Simulationen zu erreichen, insbesondere bei Proteinen mit komplexem Faltungsverhalten. Jüngere Arbeiten nutzen maschinelles Lernen, um automatisch bessere grobkörnige Kraftfelder zu entdecken, doch das Training dieser Modelle erforderte typischerweise Millionen detaillierter Schnappschüsse, jeweils mit den auf jedes Atom bezogenen Kräften — eine enorme Daten- und Rechenlast.
Physikalische Kräfte mit informativem Rauschen verbinden
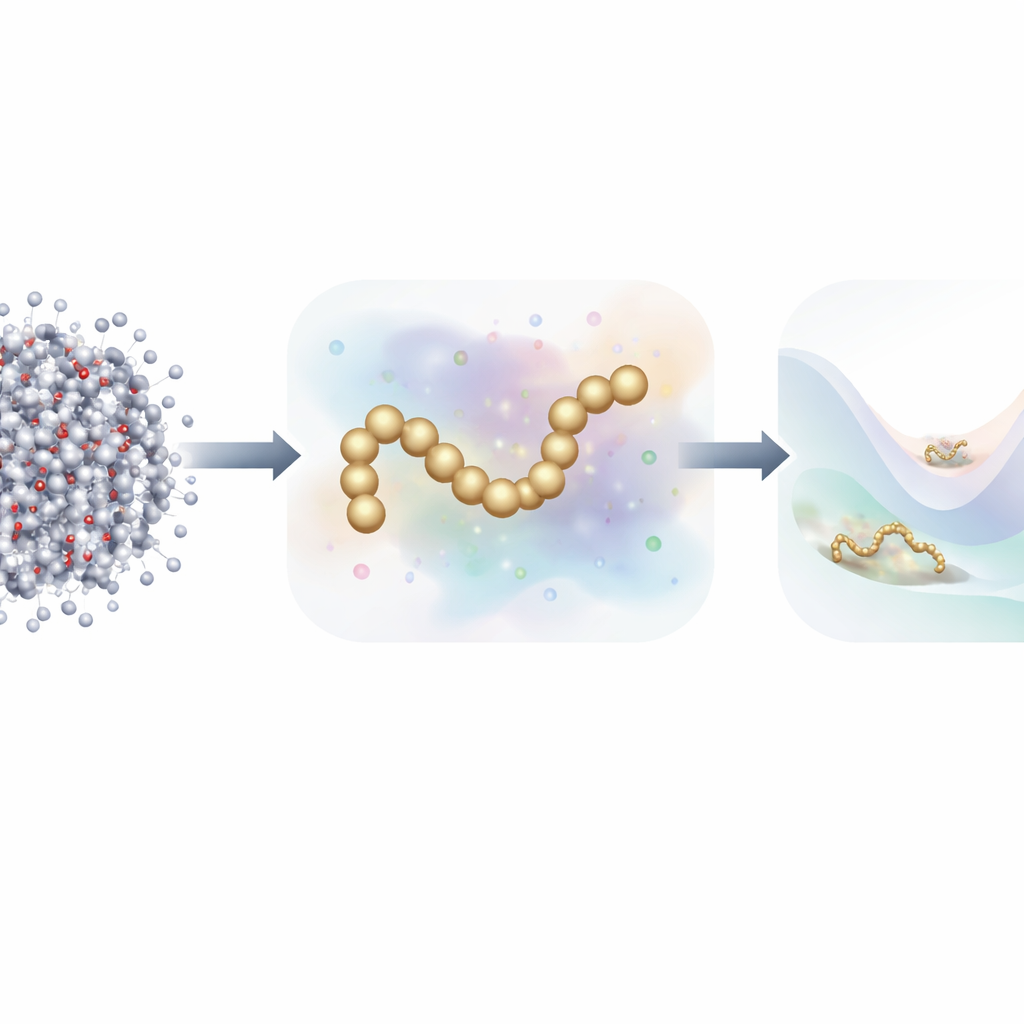
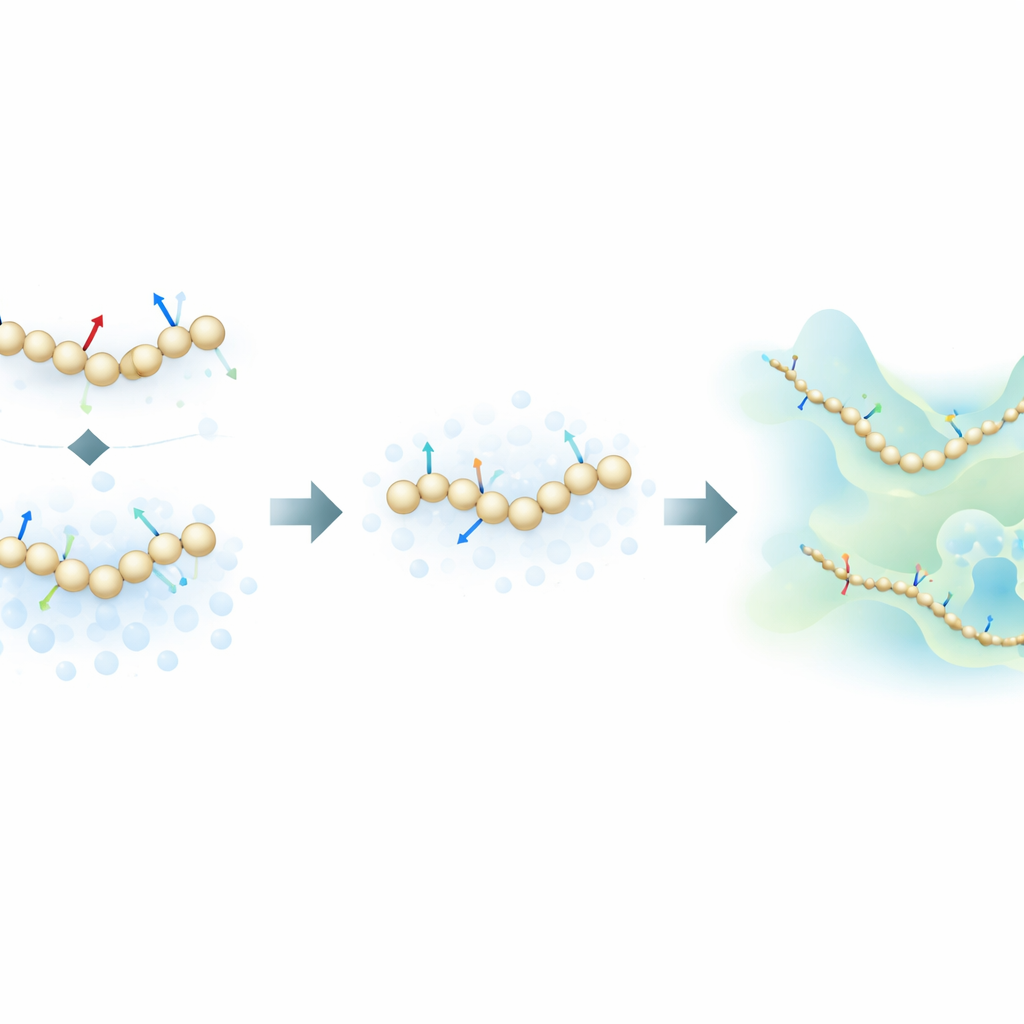
Die Autoren schlagen eine neue Trainingsstrategie vor, die sich an generativen Diffusionsmodellen orientiert — derselben Algorithmusklasse, die hinter vielen modernen KI-Bildgeneratoren steht. Anstatt nur aus den in atomistischen Simulationen berechneten physikalischen Kräften zu lernen, nutzt ihre Methode zusätzlich Informationen darüber, wie molekulare Strukturen im Raum verteilt sind, indem kontrolliert Rauschen zu grobkörnigen Konfigurationen hinzugefügt wird. In diesem Rahmen ist Rauschen nicht nur eine Störgröße, die entfernt werden muss; es wird zu einer zusätzlichen Informationsquelle. Durch die mathematische Vereinheitlichung des traditionellen Ansatzes des „Force Matching“ mit Denoising‑Techniken aus Diffusionsmodellen kann die Methode die zugrundeliegende Energielandschaft eines Proteins mit deutlich weniger gelabelten Beispielen erschließen.
Einfachen Modellen beibringen, komplexe Proteine zu imitieren
Um ihre Idee zu prüfen, trainierten die Forschenden neuronale grobkörnige Modelle für mehrere Proteine zunehmender Komplexität: die kleinen Miniproteine Chignolin und Trp‑Cage, das etwas größere NTL9 und das 76‑Restprotein Ubiquitin. Sie verglichen drei Trainingsmodi: nur atomistische Kräfte, nur rauschbasierte Informationen und eine Kombination beider. Für die kleineren Proteine zeigten sie, dass der neue kombinierte Ansatz die wesentlichen Merkmale der Faltungslandschaft reproduzieren kann — wie die relative Stabilität gefalteter und ungefalteter Zustände sowie das Vorkommen von Zwischenzuständen — und dabei bis zu Hundertmal weniger Trainingsdaten benötigt als herkömmliche Force‑Matching‑Methoden. Überraschenderweise erreichten in datensparsamen Szenarien selbst Modelle, die ausschließlich mit rauschbasierten Informationen trainiert wurden, oft eine Genauigkeit, die mit oder besser war als die reine Kraft‑basierte Ausbildung.
Größere und schwierigere Proteinsysteme erreichen
Ubiquitin stellt einen anspruchsvolleren Test dar: Das Erfassen seines Faltens und Entfaltens bei realistischen Temperaturen erforderte historisch spezialisiertes Gerät und extrem lange atomistische Läufe. Hier trainieren die Autoren grobkörnige Modelle mit einem moderaten Datensatz, der kurze Gleichgewichtssimulationen um den gefalteten Zustand sowie nicht‑gleichgewichtige ‚Pulled‘‑Simulationen enthält, bei denen das Protein gezwungen gedehnt wird. Trotz dieses verzerrten Trainingssatzes und des Fehlens einer perfekten atomistischen Referenz unter denselben Bedingungen rekonstruiert das mit Kräften und Rauschen kombinierte Modell ein realistisches Bild, in dem gefaltete und ungefaltete Zustände koexistieren, wobei der gefaltete Zustand stabilitätsmäßig bevorzugt ist. Dagegen stabilisiert ein nur mit Kräften trainiertes Modell den gefalteten Zustand gar nicht, während ein nur auf Rauschen basierendes Modell ungefaltete Strukturen bevorzugt. Bemerkenswert ist, dass keines der grobkörnigen Modelle einfach die extrem gestreckten Formen aus den Trainingsdaten auswendig übernimmt, was darauf hindeutet, dass die gelernte Energielandschaft physikalisch sinnvoll ist und nicht nur ein Abdruck der Eingangstrajektorien.
Was das für zukünftige Simulationen bedeutet
Indem Rauschen zu einem Trainingssignal gemacht und mit physikalischen Kräften verschmolzen wird, zeigt diese Arbeit, dass genaue grobkörnige Modelle von Proteinen aus deutlich kleineren und weniger idealen Datensätzen gebaut werden können als bisher angenommen. Praktisch bedeutet das, dass Forschende womöglich keine millisekundenlangen atomistischen Simulationen auf spezialisierten Supercomputern mehr benötigen, bevor sie das Verhalten eines Biomoleküls mit maschinell gelernten grobkörnigen Dynamiken untersuchen können. Stattdessen könnten bescheidenere Simulationen auf allgemein verfügbaren Rechnern ausreichen, um leistungsfähige reduzierte Modelle zu trainieren, die wichtige Faltungspfade und thermodynamische Verhältnisse erfassen. Zwar bleiben Fragen offen, etwa wie man das hinzugefügte Rauschen am besten auswählt und interpretiert und wie die Methode bei noch größeren, komplexeren Biomolekül‑Assemblies funktioniert, doch dieser Ansatz senkt die Hürde, datengetriebene grobkörnige Simulationen routinemäßig in der Molekülwissenschaft einzusetzen, erheblich.
Zitation: Durumeric, A.E.P., Chen, Y., Pasos-Trejo, A.S. et al. Learning data-efficient coarse-grained molecular dynamics from forces and noise. Nat Commun 17, 2493 (2026). https://doi.org/10.1038/s41467-026-70818-0
Schlüsselwörter: großkörnige molekulardynamik, maschinelles Lernen Kraftfelder, Proteinfaltungssimulationen, Diffusionsmodelle in der Chemie, dateneffiziente Simulation