Clear Sky Science · de
Eine pathogene Tau‑Mutation verursacht Störungen des Autophagie‑Lysosom‑Systems und begrenzt den Tau‑Abbau in einem Modell der frontotemporalen Demenz
Wenn die Aufräumtruppen des Gehirns nicht mehr nachkommen
Warum entwickeln manche Menschen Jahrzehnte vor dem hohen Alter verheerende Gedächtnis‑ und Verhaltensstörungen? Diese Studie geht dieser Frage nach, indem sie sich auf ein einzelnes Hirnprotein, Tau, und die winzigen zellulären „Recyclingzentren“ konzentriert, die es normalerweise in Schach halten. Durch Beobachtung lebender menschlicher Nervenzellen unter sehr scharfen Mikroskopen zeigen die Forschenden, wie eine krankheitsverursachende Tau‑Mutation das zelluläre Entsorgungssystem verstopft und wie die Anregung dieses Systems mit einem kleinen Molekül helfen kann, das Durcheinander zu beseitigen. Ihre Ergebnisse könnten auf neue Behandlungsstrategien für bestimmte Formen der Demenz hinweisen.
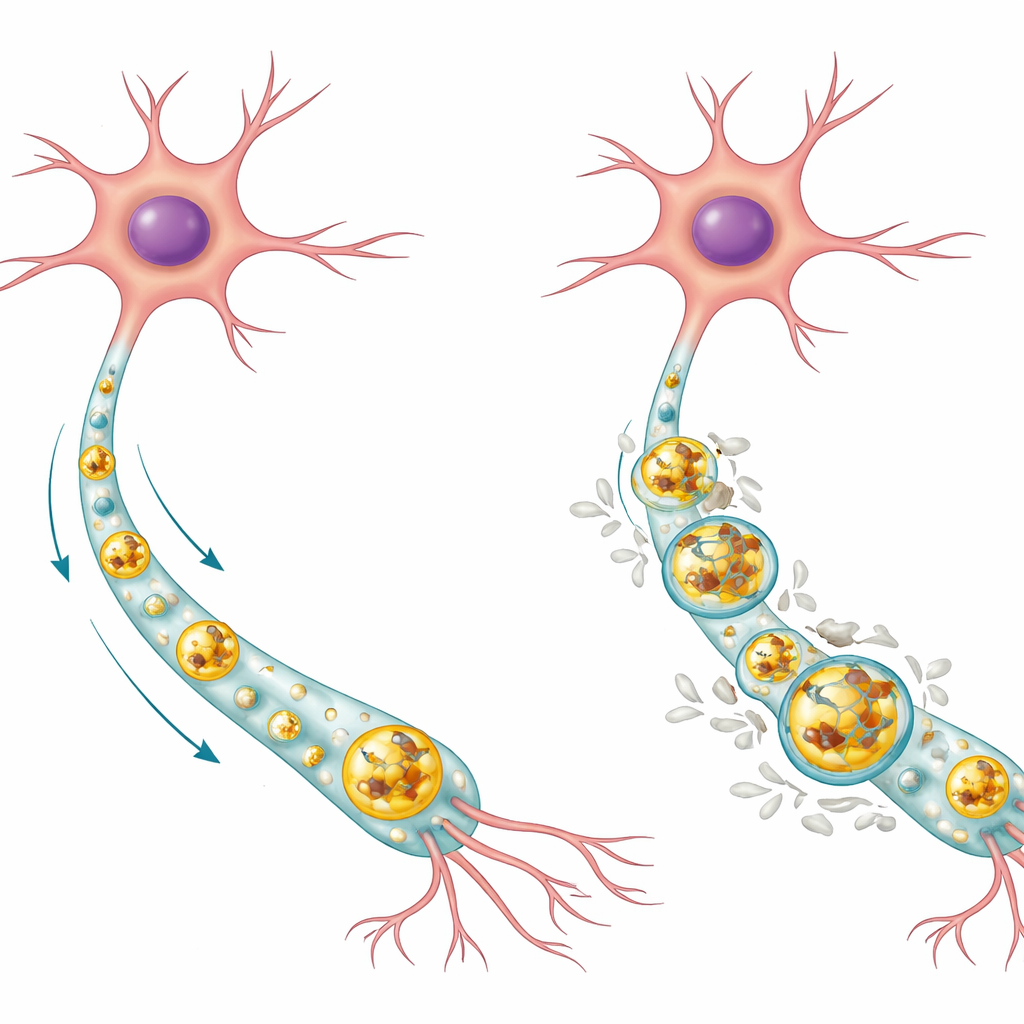
Wie Nervenzellen normalerweise den Müll entsorgen
Neuronenzellen sind langlebig und können beschädigtes Material nicht einfach durch Teilung verdünnen, daher sind sie stark auf interne Aufräumsysteme angewiesen. Ein zentraler Weg ist der Autophagie‑Lysosom‑Pfad. Dabei werden unerwünschte Proteine und verschlissene Zellbestandteile in membranumhüllte Säckchen, sogenannte Autophagosomen, eingeschlossen, die dann mit enzymgefüllten Kompartimenten – den Lysosomen – verschmelzen, wo die Fracht abgebaut und recycelt wird. In gesunden menschlichen Neuronen fanden die Autoren, dass normales Tau dazu neigt, sich im sauren Inneren der Lysosomen anzusammeln, wo es abgebaut werden kann, während die phosphorylierte Form von Tau (eine chemische Modifikation, die mit Krankheit in Verbindung gebracht wird) eher auf der Außenseite der Lysosomen sitzt. Die meisten Lysosomen in gesunden Zellen waren ganz ohne Tau, was darauf hindeutet, dass dieses System Tau‑Spiegel normalerweise niedrig und gut kontrolliert hält.
Was in einer genetischen Form der Demenz schiefgeht
Das Team konzentrierte sich auf eine Mutation im MAPT‑Gen, genannt p.R406W, die eine vererbte Form der frontotemporalen Demenz verursacht und Alzheimer‑ähnlichen Gedächtnisverlust nachahmen kann. Mithilfe von Stammzelltechnologie programmierten sie Hautzellen von Patientinnen und Patienten in induzierte pluripotente Stammzellen um und differenzierten diese dann in große Mengen menschlicher Neuronen, die entweder die Mutation trugen oder mittels Geneditierung wieder normalisiert worden waren. In den mutierten Neuronen waren Gesamt‑Tau und phosphoryliertes Tau deutlich höher, nicht weil die Zellen mehr Tau produzierten, sondern weil sie es weniger effizient entfernten. Superauflösungsbilder zeigten, dass nahezu alle Lysosomen in den mutierten Zellen mit Tau gefüllt waren und insbesondere die phosphorylierte Form Tau die Lysosomenmembran überzog. Diese Anhäufung deutete darauf hin, dass der wesentliche Protein‑Entsorgungsweg der Zelle verstopft war.
Verstopfte Recyclingzentren und stockender Verkehr
Bei genauerem Hinsehen auf die Recyclingmaschinerie stellten die Forschenden fest, dass Lysosomen in den mutierten Neuronen zahlreicher, größer waren und dazu neigten, weiter vom Zellkörper entfernt zu liegen. Live‑Aufnahmen mit fluoreszierenden Farbstoffen zeigten, dass diese Lysosomen sich langsamer bewegten und kürzere Strecken entlang der Nervenfasern zurücklegten, obwohl die zugrunde liegenden Mikrotubulus‑Spurwege normal erschienen. Die mutierten Neuronen enthielten außerdem mehr Autophagosomen, mehr des Fracht‑Adapterproteins p62 und zusätzliche Lipidtröpfchen — Anzeichen dafür, dass Material zur Entsorgung markiert, aber nicht vollständig abgebaut wurde. Mit einem pH‑sensitiven Reporter fanden sie, dass Autophagosomen in den mutierten Zellen häufig nicht richtig mit Lysosomen verschmolzen, was zu einem Aufstau von „halb fertigen“ Recyclingvesikeln und zu weitreichenden Defekten der zellulären Reinigung führte, nicht nur für Tau, sondern auch für andere Fracht.
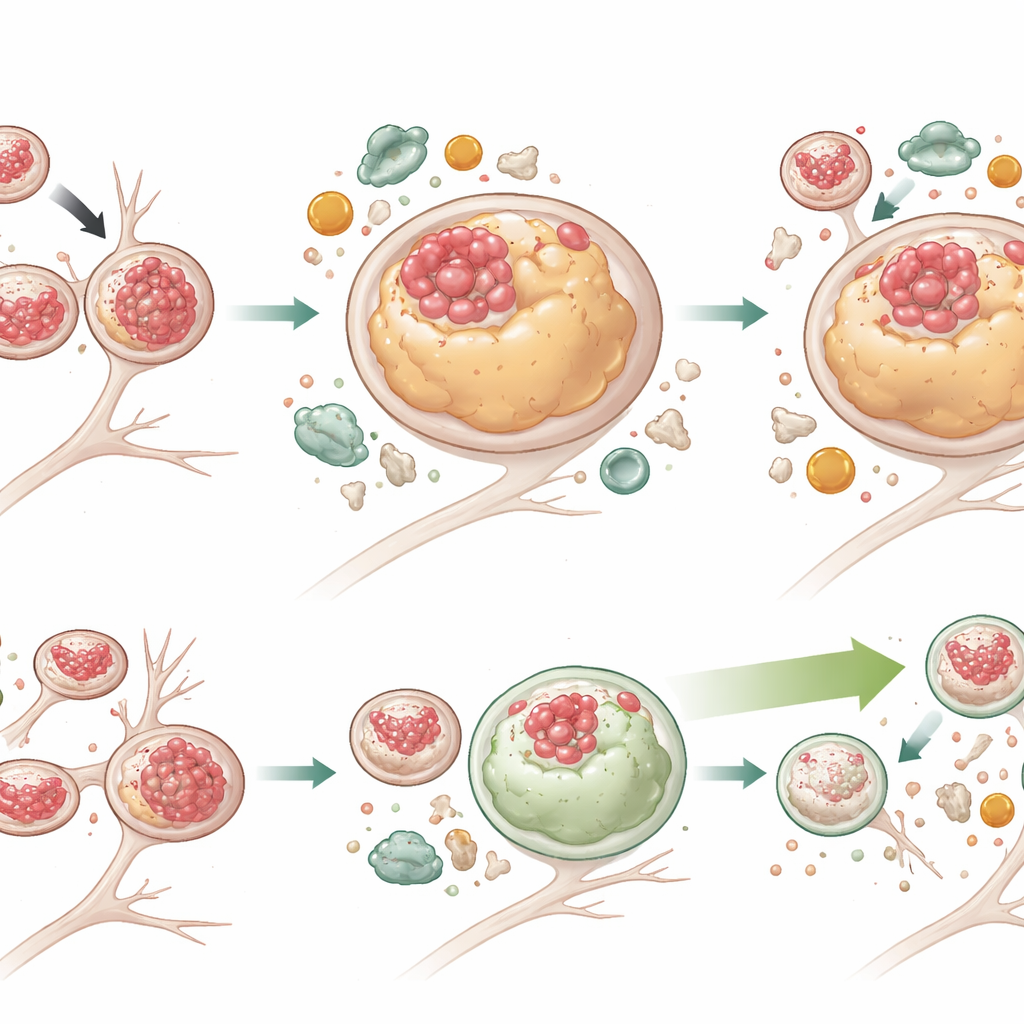
Das Aufräumen ankurbeln, ohne den Stau zu beheben
Um zu prüfen, ob die Verstärkung der Autophagie diese Probleme überwinden kann, behandelte das Team Neuronen mit G2‑567, einem kleinen Molekül, das zuvor gezeigt wurde, dass es das Autophagie‑Lysosom‑System stimuliert. Nach zweiwöchiger Behandlung hatten die mutierten Neuronen deutlich niedrigere Spiegel sowohl von Gesamt‑Tau als auch von phosphoryliertem Tau, und viele Lysosomen waren wieder frei von Tau. Die Lysosomen schrumpften außerdem wieder in Richtung Normalgröße. Marker aktiver Autophagie stiegen an, während p62 — ein Indikator für blockierten Abbau — in den mutierten Zellen abnahm, was einen effektiveren Frachtabbau zeigte. Interessanterweise korrigierte G2‑567 nicht alle Defekte: Lysosomen in den mutierten Neuronen blieben tendenziell weiter vom Zellkörper entfernt und bewegten sich träge, und ein mit Lysosomtransport verknüpftes Adapterprotein (JIP3) blieb erhöht. Das legt nahe, dass Bewegungs‑ und Abbau‑Funktionen der Lysosomen teilweise entkoppelt sein können und dass die alleinige Verbesserung des Abbaus ausreichen könnte, um toxische Tau‑Akkumulation zu reduzieren.
Was das für künftige Demenzbehandlungen bedeutet
Für Laien ist die wichtigste Erkenntnis, dass in diesem genetischen Modell der frontotemporalen Demenz das Problem nicht einfach darin besteht, dass Tau abnormal wird; vielmehr kommt das Recycling‑System der Neuronen nicht nach. Die p.R406W‑Tau‑Mutation stört mehrere Schritte des Autophagie‑Lysosom‑Wegs direkt, wodurch Tau — insbesondere seine phosphorylierte Form — auf und in Lysosomen sowie andere nicht abgebauten Materialien akkumuliert. Durch pharmakologisches Anstoßen der zellulären Aufräummaschinerie konnten die Forschenden die Tau‑Spiegel senken und die Lysosomgröße normalisieren, obwohl Transportdefekte fortbestanden. Diese Ergebnisse untermauern die Idee, dass Medikamente, die sicher Autophagie und lysosomale Funktion steigern, helfen könnten, das Protein‑Gleichgewicht bei Tau‑bedingten Demenzen und vielleicht auch bei häufigeren Erkrankungen wie der Alzheimer‑Krankheit wiederherzustellen.
Zitation: Mirfakhar, F.S., Marsh, J.A., Sato, C. et al. A pathogenic Tau mutation drives autophagy-lysosome dysfunction that limits Tau degradation in a model of frontotemporal dementia. Nat Commun 17, 2699 (2026). https://doi.org/10.1038/s41467-026-70473-5
Schlüsselwörter: Tau‑Protein, Autophagie, Lysosom‑Dysfunktion, frontotemporale Demenz, Neurodegeneration