Clear Sky Science · de
DiNovo ermöglicht hochgradige Abdeckung und hohe Zuverlässigkeit bei der de-novo-Peptidsequenzierung durch Spiegel-Proteasen und Deep Learning
Proteine in neuer Detailtiefe sichtbar machen
Proteine sind die winzigen Maschinen, die unsere Zellen am Leben erhalten, doch ihre vollständige Entschlüsselung der Bausteine ist weiterhin überraschend schwierig. Dieses Paper stellt DiNovo vor, ein neues Software‑System, das Forschenden hilft, Proteinfragmente deutlich vollständiger und zuverlässiger zu „lesen“ als bisher. Durch die Kombination eines cleveren biochemischen Tricks mit moderner künstlicher Intelligenz verspricht es, bislang verborgene Proteine, Krankheitsmarker und sogar Immunziele aufzudecken, die traditionelle Methoden oft übersehen.
Warum das Lesen von Proteinfragmenten so schwierig ist
Die meisten Proteinanalysen beruhen heute darauf, Proteine in kleinere Stücke, sogenannte Peptide, zu schneiden und anschließend ihre Fragmente in einem Massenspektrometer zu wiegen. Aus diesen Massen versuchen Computer, die ursprüngliche Peptidsequenz zu rekonstruieren — ähnlich dem Lösen eines Kreuzworträtsels aus Teilhinweisen. Bestehende Methoden gehen meist davon aus, dass Peptide aus bekannten Proteindatenbanken stammen. Das funktioniert gut für vertraute Proteine, hat aber Probleme bei neuen oder unerwarteten Varianten. Die sogenannte de-novo-Sequenzierung umgeht diese Einschränkung, indem sie versucht, Peptide direkt aus den Daten zu lesen, scheitert aber häufig, weil manche Fragmente fehlen und manche Peptide nie sauber geschnitten werden.
Spiegel-Enzyme verwenden, um Lücken zu schließen
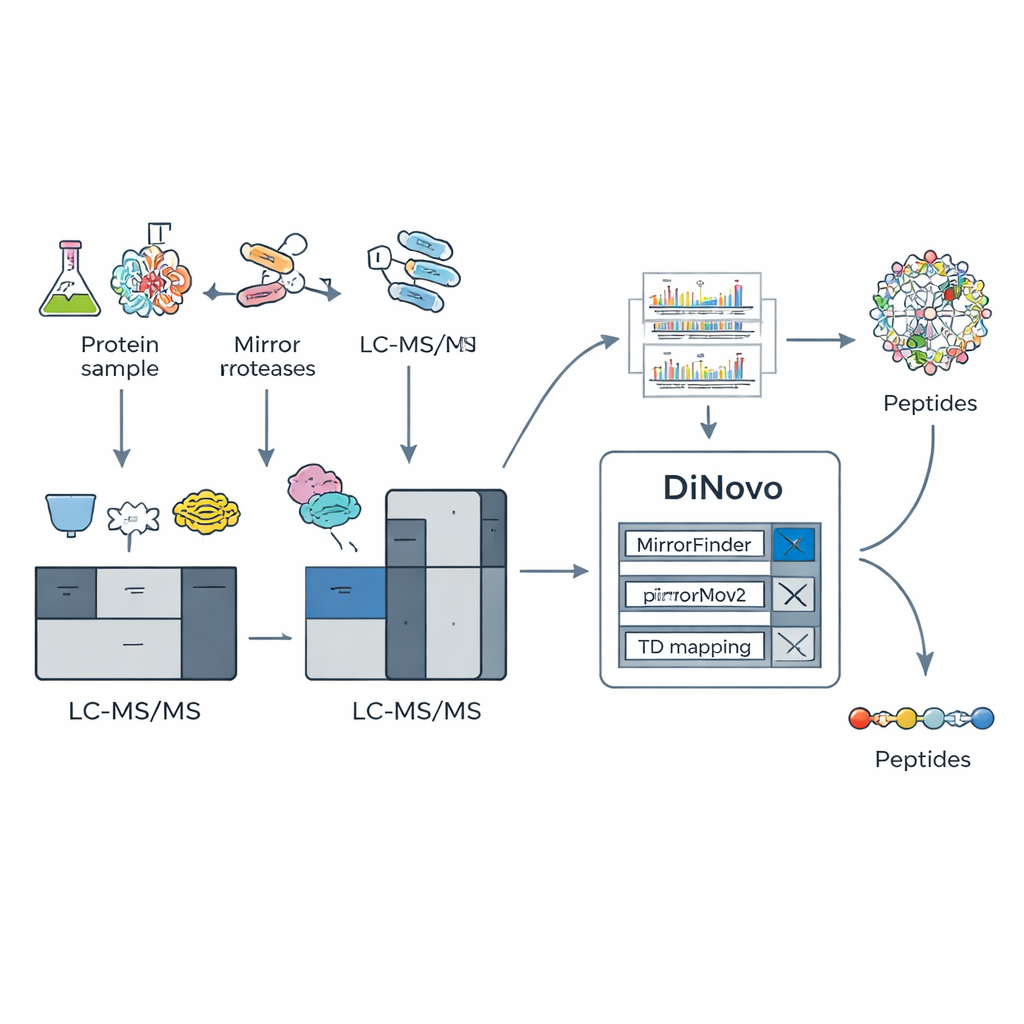
Die Kernidee von DiNovo ist die Verwendung von Paaren „Spiegel‑Proteasen“ — Schneidenzymen, die Proteine auf gegenüberliegenden Seiten desselben Aminosäurerests spalten. Zum Beispiel schneidet ein Enzym direkt vor einem Lysin, während sein Partner direkt nach diesem Lysin schneidet. So entstehen zwei verwandte Peptide, die das gleiche Innensegment teilen, aber unterschiedliche Enden besitzen. Werden diese "Spiegel"‑Peptide analysiert, enthalten ihre Massenspektren komplementäre Fragmentmuster: Was in einem Spektrum fehlt, taucht oft im anderen auf. Die Autoren zeigen, dass die Kombination solcher Spiegelpaare die Fragmentabdeckung nahezu vollständig machen kann — etwa 98 % der möglichen Schnittstellen werden durch echte experimentelle Signale unterstützt, deutlich mehr als bei Verwendung eines einzelnen Enzyms.
Eine intelligente Software‑Pipeline für Spiegel‑Daten
Um diesen biochemischen Trick nutzbar zu machen, bauten die Forscher DiNovo als End‑to‑End‑Softwareworkflow. Zuerst werden Proteine aus Bakterien und Hefe mit zwei Spiegelpaaren von Enzymen verdaut, und die resultierenden Peptide mittels hochauflösender Massenspektrometrie analysiert. DiNovo nutzt dann ein Modul namens MirrorFinder, das automatisch erkennt, welche Spektrenpaare von Spiegelpeptiden stammen — und das direkt aus den Signalmustern, ohne vorherige Sequenzannahmen. Anschließend interpretiert die Haupt‑de‑novo‑Engine MirrorNovo diese gepaarten Spektren mit Deep Learning, während eine Backup‑Graph‑Engine, pNovoM2, eine schnellere CPU‑only‑Option bietet. Zusammen übersetzen diese Werkzeuge Peaks in Aminosäuresequenzen und untersuchen zudem einzelne Spektren, die keine offensichtlichen Paare bildeten, um möglichst viele Informationen zu gewinnen.
Vertrauensmessung ohne Rückgriff auf alte Datenbanken
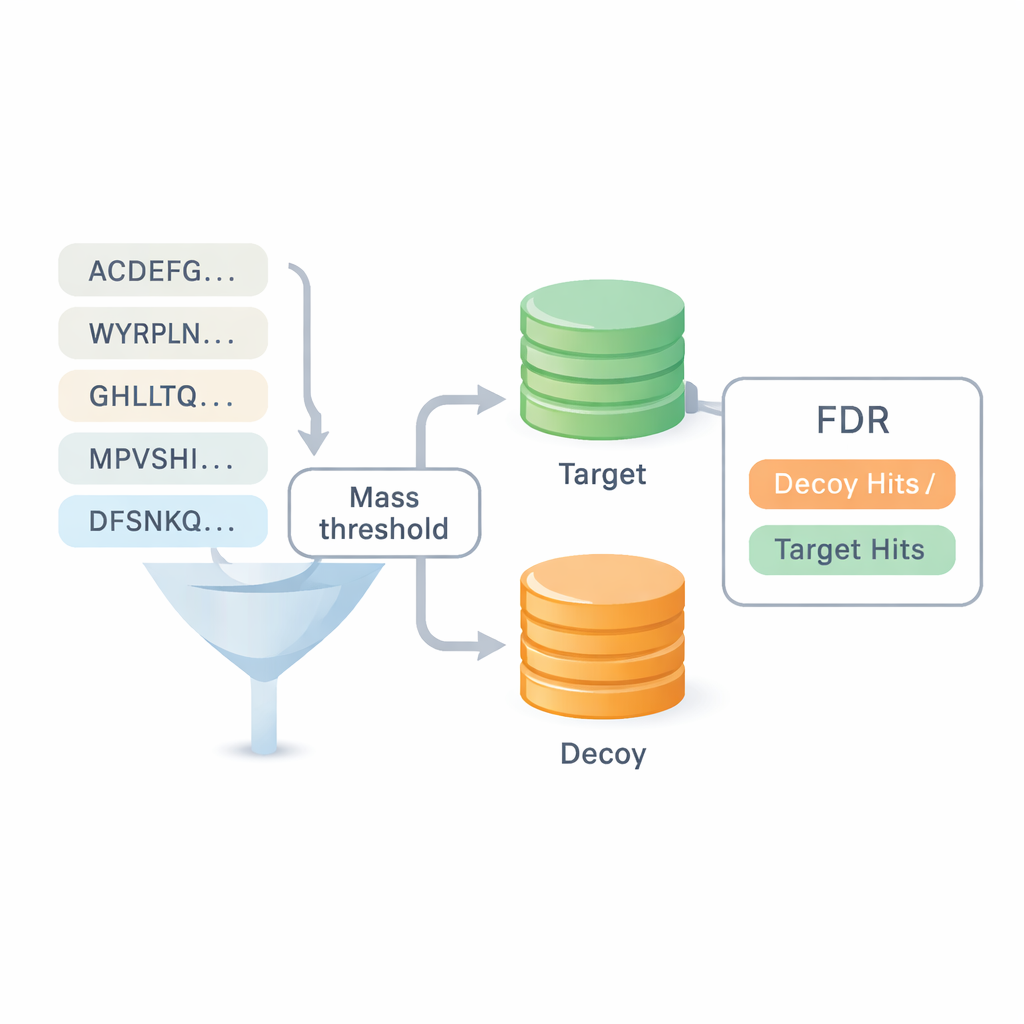
Eines der größten Probleme der de‑novo‑Sequenzierung ist, wie sehr man den Ergebnissen trauen kann. Die meisten bestehenden Benchmarks recyclen Antworten aus Datenbanksuchen, was die Grenze zwischen den Ansätzen verwischt und Fehler verbergen kann. DiNovo führt eine andere Qualitätssicherungs‑Methode namens Target‑Decoy‑Mapping ein. Hier werden die neu gelesenen Peptide gegen eine kombinierte Sammlung aus echten (Target) und künstlich vertauschten (Decoy) Proteinsequenzen abgebildet. Indem verglichen wird, wie oft Peptide in der echten Menge gegenüber der vertauschten Menge landen, kann die Software eine Fehlerrate bzw. False‑Discovery‑Rate schätzen, ohne sich auf vorherige Identifikationen zu stützen. So lässt sich DiNovo direkt mit Standard‑Datenbanksuchprogrammen unter denselben Fehlerkontrollen vergleichen.
Was DiNovo in der Praxis liefert
In Tests an Bakterien‑, Hefe‑ und Antikörperproben las DiNovo konsequent deutlich mehr Peptide und Aminosäuren als bekannte de‑novo‑Werkzeuge, die nur ein einzelnes Enzym verwenden. Mit zwei Spiegelpaaren erzeugte es zwei‑ bis dreimal mehr hochvertraute Aminosäuren als eine klassische Trypsin‑nur‑Konfiguration und identifizierte mehr Proteine bei ähnlichen Fehlerraten. Im direkten Vergleich mit drei führenden Datenbanksuchengines fand DiNovo eine ähnliche Anzahl an Aminosäuren und Proteinen, und die meisten seiner Sequenzen stimmten mit jenen der Suchengines auf denselben Spektren überein. Die Autoren argumentieren, dass dieses Abdeckungs‑ und Übereinstimmungsniveau bedeutet, dass die de‑novo‑Sequenzierung, lange Zeit als Reserveverfahren betrachtet, nun gleichberechtigt neben der Datenbanksuche stehen kann — und in manchen Fällen überlegen ist.
Großes Bild: Auf dem Weg zu vollständigem, unbeeinflusstem Proteinlesen
Für Nicht‑Spezialisten lautet die Kernbotschaft: DiNovo macht es deutlich einfacher, Proteinfragmente genau zu lesen, ohne auf vorhandene Referenzdatenbanken beschränkt zu sein. Indem die Menge gut unterstützter Sequenzinformationen verdoppelt oder verdreifacht und eigene Fehlerprüfungen bereitgestellt werden, eröffnet dieser Ansatz die Möglichkeit, unbekannte Proteine zu entdecken, feine Variationen nachzuverfolgen und komplexe Gemische zu untersuchen, in denen viele Komponenten noch unbekannt sind. Kurz: Durch die Kombination von Spiegel‑Enzymen mit Deep Learning und sorgfältiger Statistik hilft DiNovo dabei, verrauschte Spektrenspuren in ein klareres, verlässlicheres Bild der Proteine zu verwandeln, die Gesundheit und Krankheit zugrunde liegen.
Zitation: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
Schlüsselwörter: Proteomik, de-novo-Peptidsequenzierung, Massenspektrometrie, Deep Learning, Spiegel-Proteasen