Clear Sky Science · de
Die gezielte Hemmung der de‑novo‑Pyrimidinsynthese macht gegenüber PARP‑Inhibitoren resistente Ovarialkarzinome anfällig für kupfervermittelte ATR‑Inaktivierung
Warum diese Forschung wichtig ist
Viele Frauen mit Eierstockkrebs werden mit Medikamenten behandelt, die die Fähigkeit von Tumorzellen sabotieren, beschädigte DNA zu reparieren. Diese Wirkstoffe, PARP‑Inhibitoren genannt, können anfangs gut wirken, doch Tumoren passen sich häufig an und wachsen zurück. Diese Studie zeigt, wie ein kupfertransportierendes Medikament und eine zentrale metabolische Schwäche resistente Eierstockkarzinome über ihre Belastungsgrenze bringen können. Das weist auf intelligentere Kombinationsbehandlungen und länger anhaltende Antworten hin.
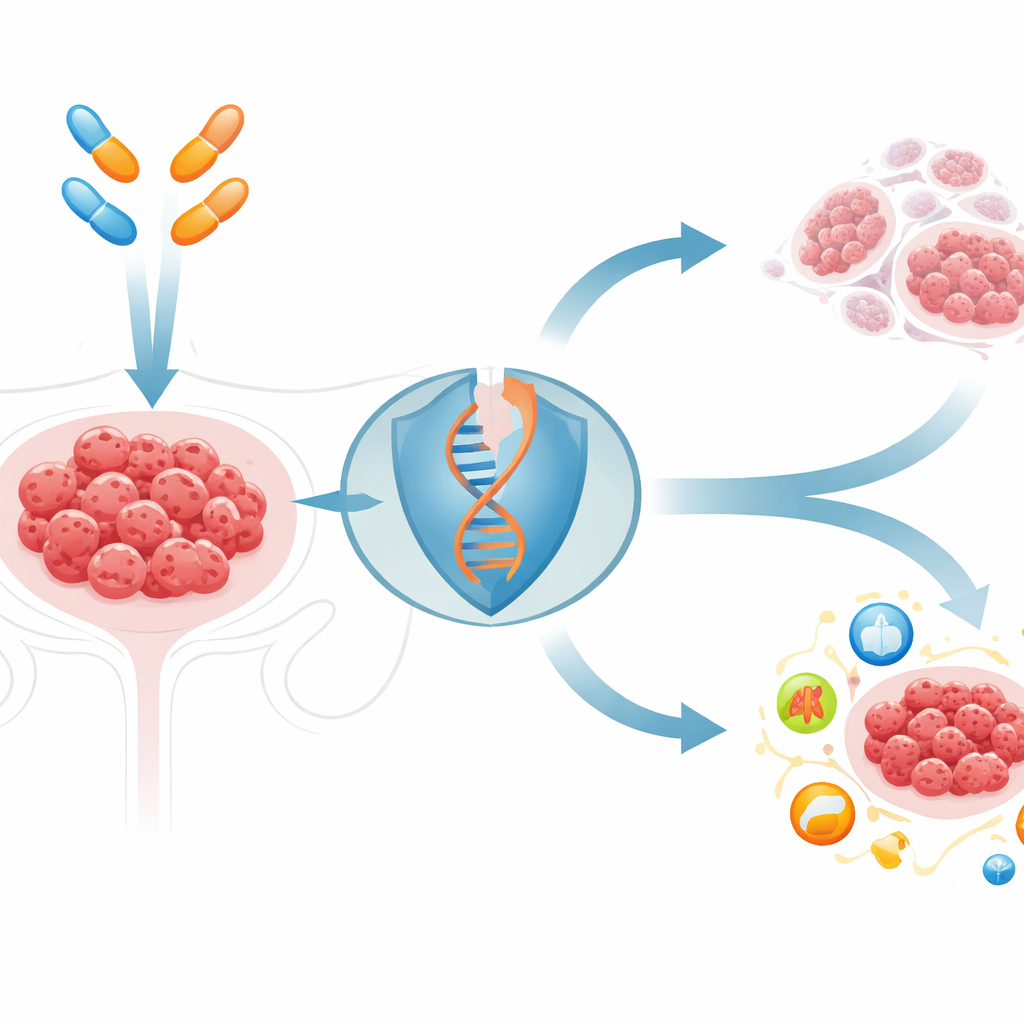
Die hartnäckigen Tumorverteidigungen knacken
PARP‑Inhibitoren nutzen eine Schwäche im DNA‑Reparaturmechanismus mancher Krebsarten aus. Am besten wirken sie in Tumoren mit erblichen BRCA‑Defekten, doch die meisten Eierstockkarzinome haben intakte BRCA‑Gene und sprechen schlecht oder nur kurz an. Die Forschenden testeten 144 zelltodbezogene Verbindungen zusammen mit einem Standard‑PARP‑Inhibitor und fanden, dass ein Medikament, Elesclomol, herausstach. Elesclomol transportiert Kupfer in Zellen. In Kombination mit PARP‑Inhibitoren in BRCA‑intakten Eierstockkrebszellen und Mausmodellen erhöhte dieses kupfersteigernde Mittel die DNA‑Schäden deutlich und verkleinerte Tumoren deutlich mehr als eines der beiden Medikamente allein, ohne offensichtliche Toxizität in gesunden Organen.
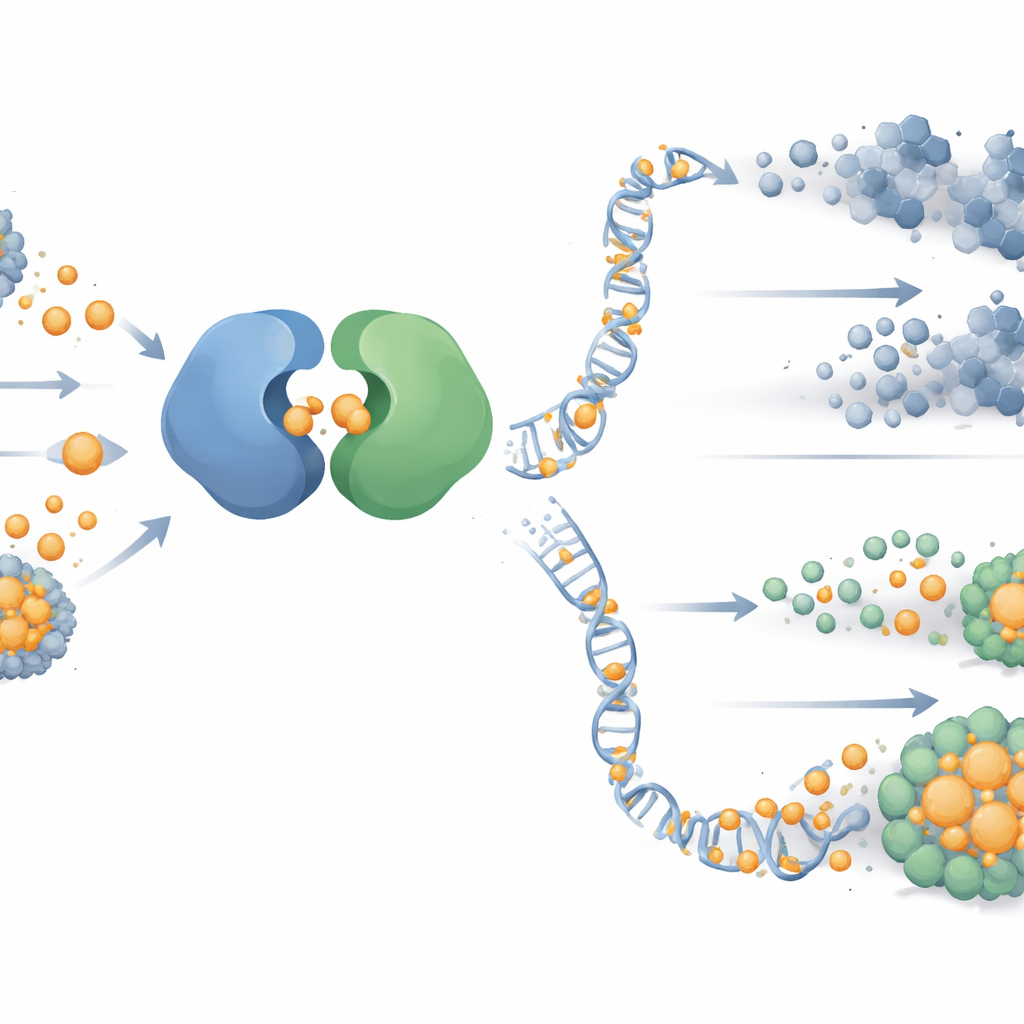
Kupfer blockiert einen DNA‑Reparaturschalter
Um zu verstehen, warum Kupfer die PARP‑Blockade tödlicher machte, untersuchten die Forschenden einen wichtigen DNA‑Schadens‑Signalweg, der von einem Protein namens ATR ausgeht. Dieser Weg hilft Zellen, zu überleben, wenn ihre DNA‑Replikation gestresst ist — genau die Situation, die PARP‑Inhibitoren hervorrufen. In Tumorzellen, die eine erste Medikamentenexposition überlebt hatten, waren ATR und sein Partnerprotein CHK1 stark aktiviert, während ein verwandter Weg (ATM‑CHK2) ruhig blieb. Detaillierte biochemische Tests und computergestützte Strukturmodelle zeigten, dass Kupfer direkt an ATRs Helferprotein ATRIP an bestimmten Cystein‑Stellen bindet. Diese Bindung verzerrt die Form von ATRIP, unterbricht seinen Kontakt zu ATR und schaltet das ATR‑CHK1‑Signal aus, sodass beschädigte DNA unrepariert bleibt und PARP‑behandelte Krebszellen deutlich eher sterben.
Die verborgene Rolle des Nukleotid‑Treibstoffs
Selbst wenn ATR und PARP beide ausgeschaltet waren, hielten sich einige Krebszellen und verbleibende Tumoren weiter. Um herauszufinden, wie, analysierten die Forschenden Hunderte kleiner Moleküle in medikamentenangepassten Zellen. Sie fanden einen markanten Anstieg von DNA‑Bausteinen, den Pyrimidinen, insbesondere jener, die über den de‑novo‑Weg synthetisiert werden, bei dem Zellen diese Komponenten von Grund auf neu herstellen. Tracer‑Experimente bestätigten, dass resistente Zellen mehr Stickstoff aus Glutamin in neue Pyrimidine einleiteten, während Purin‑Bausteine nicht vergleichbar erhöht waren. Das Zufügen zusätzlicher Pyrimidinbestandteile wie Uridin oder Thymin zu Kulturen schwächte die Wirksamkeit der Kombination aus PARP‑ und ATR‑ oder kupferbasierter Behandlung ab, was darauf hindeutet, dass ein reichliches Angebot an DNA‑Bausteinen Tumoren hilft, ansonsten tödliche DNA‑Schäden zu tolerieren.
Einen metabolischen Schwachpunkt treffen
Das Team prüfte dann, ob die Blockade dieser Pyrimidin‑Versorgung die Fluchtmöglichkeit schließen könnte. Sie verwendeten BAY‑2402234, ein experimentelles Medikament, das DHODH hemmt, ein Schlüsselenzym der de‑novo‑Pyrimidinsynthese. In Eierstockkrebszelllinien und patientenabgeleiteten Organoiden stellte das Hinzufügen des DHODH‑Inhibitors die Sensibilität für PARP plus ATR‑ oder Kupfer‑Behandlung wieder her und beseitigte zuvor medikamententolerante Zellen. In Mausmodellen und in acht patientenabgeleiteten Xenograft‑Modellen zeigten Tumoren, die gegenüber PARP allein — und sogar gegenüber PARP kombiniert mit ATR‑ oder Kupferblockade — resistent waren, eine starke Wachstumshemmung, wenn die Pyrimidinsynthese ebenfalls blockiert wurde. Tumoren mit von Natur aus hohen Pyrimidinmetabolit‑Spiegeln ließen sich mit PARP‑basierten Ansätzen am schwersten behandeln, sprachen aber an, wenn dieser metabolische Weg angegriffen wurde.
Was das für Patientinnen bedeuten könnte
Diese Studie offenbart zwei verknüpfte Verwundbarkeiten in gegenüber PARP‑Inhibitoren resistentem Eierstockkrebs. Erstens kann Kupfer als gezieltes Werkzeug verwendet werden, um einen wichtigen DNA‑Reparaturschalter, ATR, zu deaktivieren, indem es dessen Partnerschaft mit ATRIP trennt und Standard‑DNA‑angreifende Medikamente wirksamer macht. Zweitens werden Tumoren, die sich durch eine Hochregulierung der Pyrimidinproduktion anpassen, abhängig von diesem Stoffwechselweg, und dessen Blockade kann sie wieder empfindlich gegenüber der Behandlung machen. Praktisch gesprochen sprechen die Ergebnisse für maßgeschneiderte Kombinationsstrategien: PARP‑Inhibitoren plus ATR‑gezielte Wirkstoffe für Tumoren mit geringer Pyrimidinabhängigkeit und ein dreifaches Vorgehen, das zusätzlich die Pyrimidinsynthese blockiert, für Tumoren, die metabolisch auf Resistenz vorbereitet sind. Obwohl weitere klinische Prüfungen nötig sind, zeichnet die Arbeit eine klarere Karte zum Überwinden einer der hartnäckigsten Formen von Medikamentenresistenz beim Eierstockkrebs.
Zitation: Nan, Y., Wang, K., Hu, M. et al. Targeting de novo pyrimidine synthesis confers vulnerability to copper-mediated ATR inactivation in PARP inhibitor-resistant ovarian cancer. Nat Commun 17, 3142 (2026). https://doi.org/10.1038/s41467-026-70001-5
Schlüsselwörter: Eierstockkrebs, PARP‑Inhibitoren, Kupfertherapie, DNA‑Reparatur, Pyrimidin‑Stoffwechsel