Clear Sky Science · de
Multimodale Zerlegung der zelltypspezifischen TDP-43-Pathologie im Motorkortex
Warum diese Forschung für Menschen wichtig ist
Die amyotrophe Lateralsklerose (ALS) und die frontotemporale Demenz (FTD) sind verheerende Hirnerkrankungen, die Menschen ihre Bewegungsfähigkeit, Sprache und Persönlichkeit rauben. Die meisten ALS-Patienten und viele FTD-Patienten teilen ein gemeinsames mikroskopisches Kennzeichen: Ablagerungen eines Proteins namens TDP-43 an Stellen, an denen es normalerweise nicht vorkommt. Diese Studie stellt zwei praktische Fragen mit großen Implikationen für künftige Therapien: Welche Hirnzellen sind genau am stärksten von TDP-43-Störungen betroffen, und was geht in diesen Zellen auf der Ebene der DNA-Regulation und Genaktivität schief?
Dem Schaden in das motorische Zentrum des Gehirns folgen
Die Forschenden konzentrierten sich auf den primären Motorkortex, den Hirnbereich, der willkürliche Bewegungen steuert. Anhand postmortaler, gespendeter Hirnproben von Menschen mit ALS, ALS-FTD und neurologisch gesunden Kontrollen isolierten sie einzelne Zellkerne und bestimmten sowohl, welche Gene aktiv waren, als auch wie offen oder dicht die lokale DNA verpackt war. Dieser „multi-omische“ Ansatz, angewandt auf mehr als 180.000 Kerne, erlaubte es ihnen, Zellen in präzise Typen zu ordnen: mehrere Klassen exzitatorischer und inhibitorischer Neuronen sowie Stützzellen wie Astrozyten, Oligodendrozyten und Mikroglia. Anschließend kombinierten sie diese Daten mit räumlichen Genkartierungen aus einem anderen menschlichen Hirndatensatz, um diese Zelltypen wieder in die vertraute geschichtete Struktur des Kortex einzuordnen.
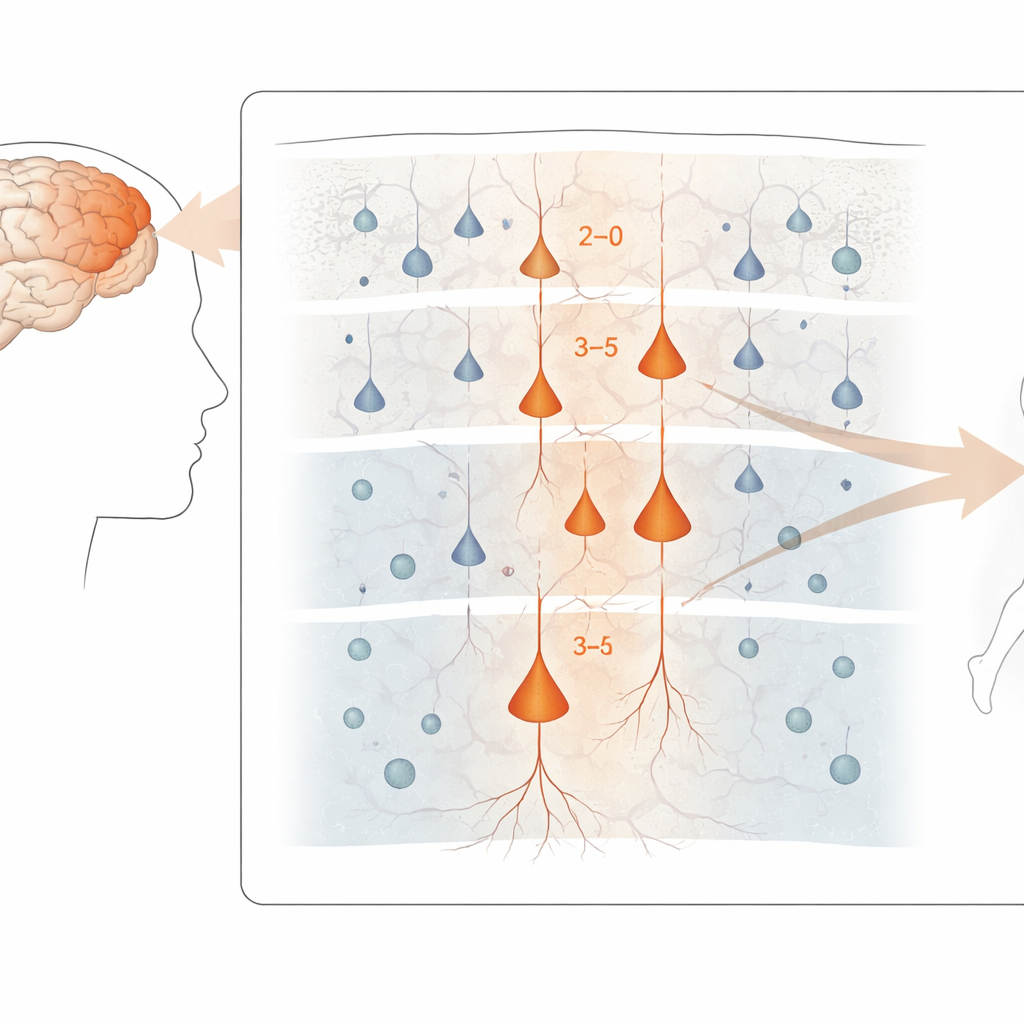
Die verwundbarsten Neurone punktgenau bestimmen
Im gesamten Motorkortex zeigten sich die stärksten krankheitsbezogenen Genveränderungen in exzitatorischen Neuronen, den Zellen, die Aktivität in den Schaltkreisen vorantreiben. Besonders betroffen waren Neurone der oberen und mittleren Schichten, die innerhalb des Kortex verbunden sind, sowie bestimmte tiefere Schichtzellen, die Signale aus dem Kortex hinaussenden — darunter die großen „Betz“-Zellen, die die spinalen Motoneurone kontrollieren. Dagegen waren inhibitorische Interneurone und viele Gliazellen auf Ebene der Genexpression weniger stark betroffen, obwohl einige von ihnen subtilere Verschiebungen zeigten. Trotz dieses molekularen Aufruhrs war das Verhältnis der Hauptzelltypen im Gewebe zwischen Patienten und Kontrollen überraschend ähnlich, was darauf hindeutet, dass die Schädigung eher die Funktion der Zellen als deren bloßen Verlust betrifft.
Wie TDP-43 die Genaktivität von innen heraus umgestaltet
Um Effekte, die direkt von TDP-43 ausgehen, von anderen Krankheitsprozessen zu trennen, verwendete das Team eine clevere Sortierstrategie. Sie markierten Kerne mit Antikörpern gegen TDP-43 und einen neuronalen Marker und trennten dann per Durchflusszytometrie Neuronen, deren Kerne TDP-43 verloren hatten (ein Zeichen der Pathologie), von solchen, die es behielten. Die Sequenzierung von mehr als 12.000 dieser Kerne zeigte, dass der Verlust von TDP-43 überwiegend in exzitatorischen Neuronen auftritt, insbesondere in bestimmten Subtypen der Schichten 2–3, 3–5, 5 und 6. In diesen verwundbaren Neuronen waren Hunderte von Genen fehlreguliert, darunter viele, die bereits mit ALS in Verbindung gebracht werden. Klassische molekulare Signaturen einer TDP-43-Fehlfunktion — wie das Auftreten „kryptischer“ zusätzlicher Stücke in STMN2- und KALRN-Transkripten und Verschiebungen bei der Stelle, an der RNA-Moleküle an ihren Enden geschnitten und polyadenyliert werden — waren in den TDP-43-defizienten Kernen deutlich angereichert.
Epigenetische Umgestaltung: nicht alle Veränderungen stammen von TDP-43
Da sie in denselben Kernen sowohl Genaktivität als auch Chromatinzugänglichkeit maßen, konnten die Autorinnen und Autoren untersuchen, welche Veränderungen mit Verschiebungen in der DNA-Verpackung einhergingen. Sie fanden Zehntausende Stellen im Genom, an denen die lokale Chromatinzugänglichkeit mit der Genexpression korrelierte. Viele der in ALS und ALS-FTD veränderten Gene lagen in solchen Regionen, was darauf hindeutet, dass ein Teil des Krankheitssignatur auf breitere epigenetische Umgestaltungen zurückzuführen ist und nicht direkt auf den Verlust von TDP-43. Interessanterweise konvergierten diese chromatinbezogenen Veränderungen häufig auf Signalwege, die an Zellkommunikation und Axonführung beteiligt sind, und sie waren besonders stark in bestimmten exzitatorischen Neuronen und Oligodendrozyten. Beim Vergleich der Genveränderungen, die mit TDP-43-Pathologie verknüpft waren, mit denen, die mit Chromatinverschiebungen assoziiert waren, zeigte sich, dass beide teilweise überlappten, aber größtenteils unterschiedliche Ebenen der Störung darstellten.
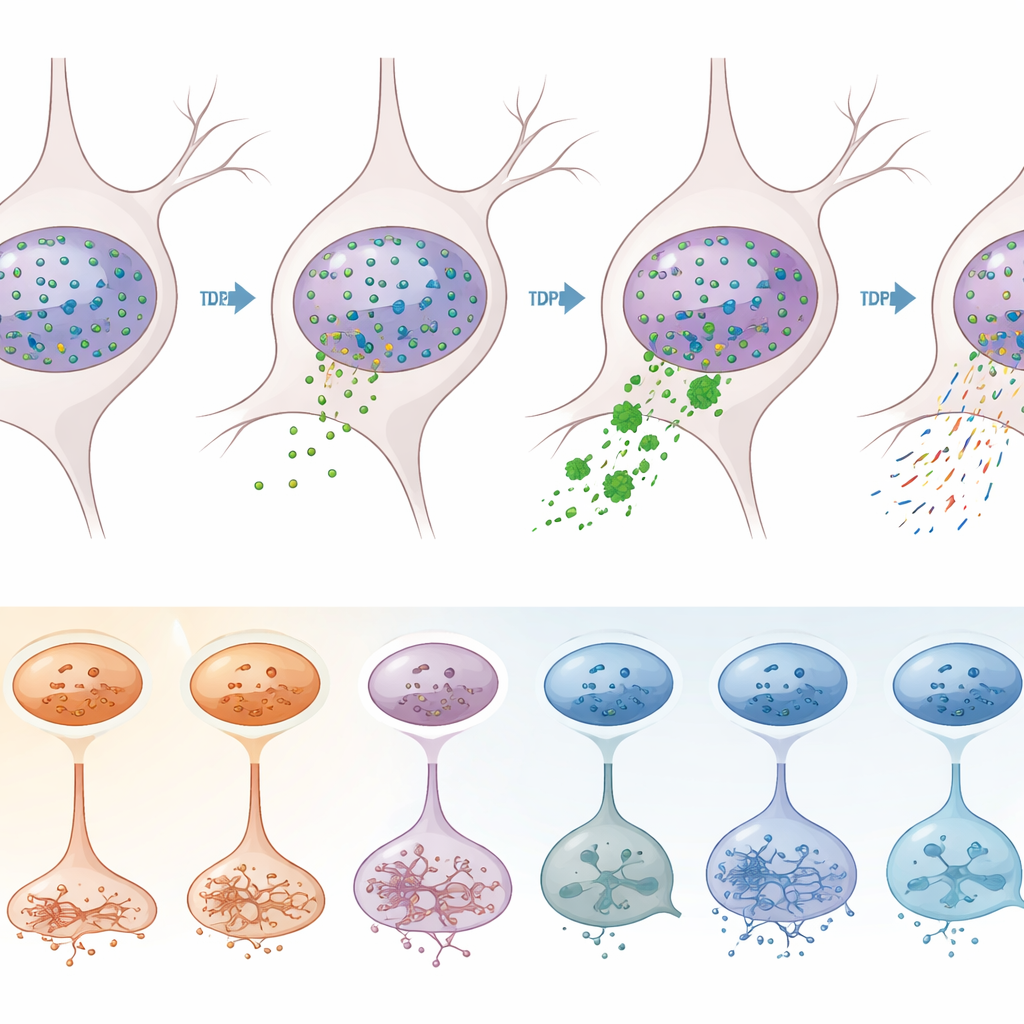
Was das für zukünftige Therapien bedeutet
Für eine allgemeine Leserschaft lautet die Kernbotschaft: ALS und ALS-FTD schädigen den Motorkortex nicht gleichmäßig. Stattdessen treffen sie bestimmte Typen exzitatorischer Neuronen und, in geringerem Maße, bestimmte Stützzellen und verändern deren Genprogramme auf Weisen, die sowohl von der Funktionsstörung von TDP-43 als auch von breiteren Veränderungen in der DNA-Verpackung und -Lesung abhängen. Diese Befunde deuten darauf hin, dass wirksame Therapien zelltypspezifisch und wegeorientiert sein müssen — zum Beispiel durch Wiederherstellung der TDP-43-Funktion oder Korrektur seiner Spleißfehler in den am stärksten verwundbaren Neuronen, während epigenetische und signalbezogene Veränderungen, die mehrere Zelltypen betreffen, separat adressiert werden. Indem die Studie diese komplexe Landschaft mit hoher Detailgenauigkeit abbildet, liefert sie eine Blaupause für die Entwicklung präziserer Interventionen, die darauf abzielen, den Verlust der Bewegungssteuerung bei ALS und ALS-FTD zu verlangsamen oder zu verhindern.
Zitation: Ruf, W.P., Kühlwein, J.K., Meier, L. et al. Multi-modal dissection of cell-type specific TDP-43 pathology in the motor cortex. Nat Commun 17, 2406 (2026). https://doi.org/10.1038/s41467-026-69944-6
Schlüsselwörter: ALS, frontotemporale Demenz, TDP-43, Neuronen des Motorkortex, Single-Nucleus-Multiomics