Clear Sky Science · de
Effiziente Stichproben großer Übergangspfade und intermediärer Konformationen in sub-mesoskopischen Proteinkomplexen
Proteine in Bewegung beobachten
Viele der Moleküle, die uns am Leben erhalten, verhalten sich weniger wie starre Lego-Steine und mehr wie winzige Maschinen, die ständig ihre Form ändern. Diese Bewegungen treiben Prozesse wie Energiegewinnung, DNA-Reparatur und das Eindringen von Viren in Zellen an. Experimente wie Kryo-Elektronenmikroskopie können inzwischen einige dieser Gestalten „einfrieren“, nicht jedoch die flüchtigen Zwischenschritte. Dieser Artikel stellt eBDIMS2 vor, eine neue Computer-Methode, die selbst für riesige molekulare Maschinen, die zuvor zu groß und zu komplex für gewöhnliche Rechner waren, die fehlenden „Frames“ der Proteinbewegung ergänzen kann.
Warum Formänderungen von Proteinen wichtig sind
Proteine verharren selten in einer einzigen Pose. Sie öffnen und schließen sich, verdrehen und biegen sich als Reaktion auf Signale wie Spannungsänderungen, pH-Wert oder die Bindung eines Partnermoleküls. Solche Verschiebungen können darüber entscheiden, ob ein Enzym aktiv ist oder nicht, oder ob ein Rezeptor ein Virus fängt oder es passieren lässt. Experimente liefern detaillierte Momentaufnahmen einiger Schlüsselformen, und molekulare Dynamik-Simulationen können diese prinzipiell verbinden, indem sie jedem Atom über die Zeit folgen. Doch die Nachverfolgung solcher Bewegungen für die riesigen Komplexe, die inzwischen in der Kryo-Elektronenmikroskopie beobachtet werden und häufig Hunderttausende bis Millionen Dalton wiegen, verlangt in der Regel Supercomputer und Wochen an Rechenzeit. Deshalb wissen wir für viele medizinisch bedeutsame Riesen noch nicht, wie ein Zustand in einen anderen übergeht.
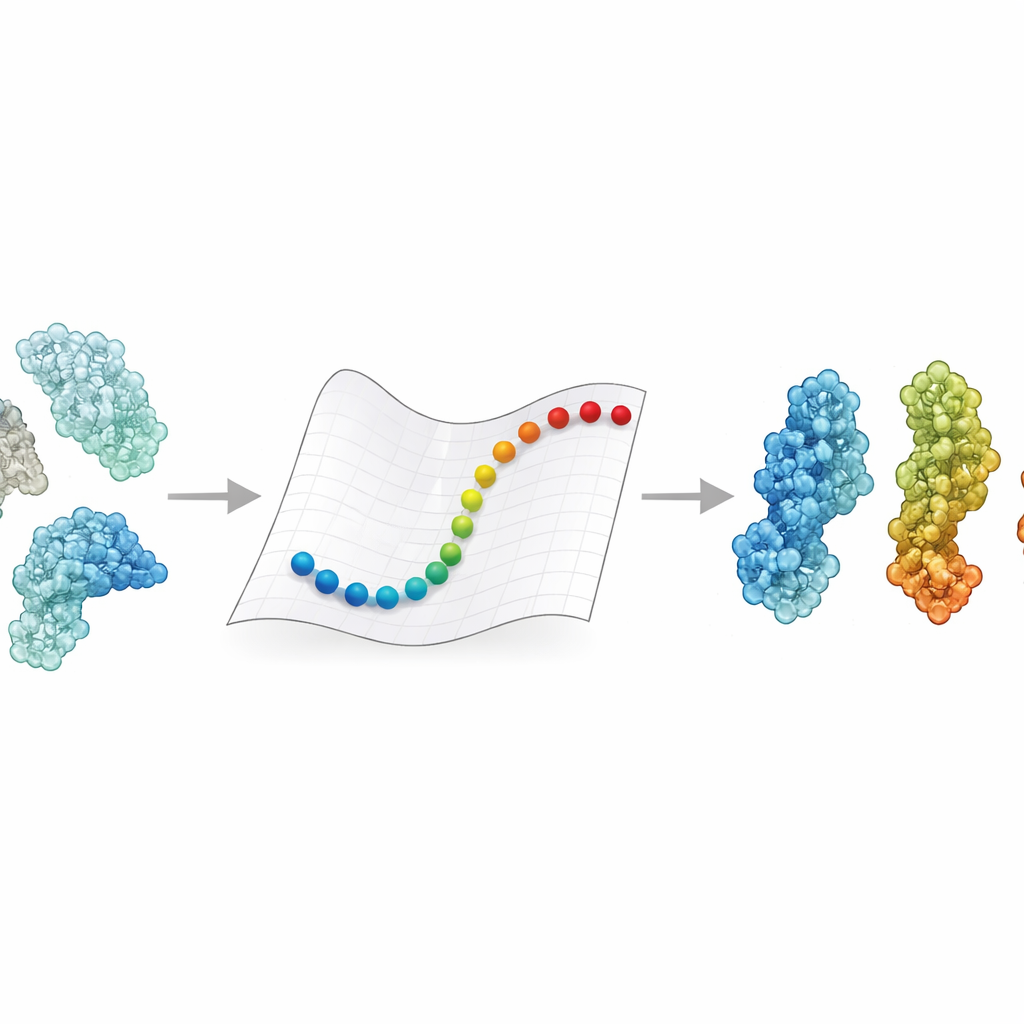
Eine schnellere Route durch Proteinlandschaften
eBDIMS2 wählt eine Abkürzung, indem es darstellt, wie Proteine repräsentiert werden und wie ihre Bewegung berechnet wird. Anstatt jedem Atom zu folgen, behandelt es jede Aminosäure als einzelnen Punkt, der in einem elastischen Netzwerk durch Federn verbunden ist. Diese Federn erfassen, wie sich verschiedene Teile des Proteins gemeinsam bewegen. Die Methode nutzt dann Brown’sche Dynamik — mathematische Regeln, die das „Zittern“ in einer Flüssigkeit nachahmen — um die Struktur von einem experimentell bekannten Zustand in Richtung eines anderen zu treiben. Entscheidend ist, dass eBDIMS2 nur Wechselwirkungen berücksichtigt, die wirklich wichtig sind, und Distanzgrenzen sowie parallele Berechnung verwendet, um die Kosten zu senken. Dadurch verbessert sich die Skalierung des Programms von ungefähr quadratisch zu nahezu linear mit der Proteingröße. In der Praxis bedeutet das, dass Übergänge für riesige Assemblies von annähernd zwei Millionen Dalton in Stunden auf einem Desktop erkundet werden können, statt de facto unerreichbar zu sein.
Die Pfade an realen Proteinen überprüfen
Um zu prüfen, ob diese schnellen Pfade biologisch sinnvoll sind, stellten die Autoren Ensembles aus 47 großen Proteinen und 15 zusätzlichen Komplexen zusammen, insgesamt Hunderte von Strukturen, meist gelöst durch Kryo-Elektronenmikroskopie. Sie nutzten die Hauptkomponentenanalyse, ein statistisches Werkzeug, das die dominanten Bewegungsmodi eines Proteins herausfiltert, um diese Strukturen in Konformationslandschaften wie offen, geschlossen, aktiv oder inaktiv zu ordnen. eBDIMS2 sollte dann Paare von Endzuständen über diese Landschaft verbinden. Die resultierenden Pfade wurden zurück in dieselben niedrigdimensionalen Karten projiziert, um zu zeigen, ob sie glatte Routen verfolgen, die in die Nähe experimentell beobachteter Zwischenstufen geraten. In mehr als 30 % der Systeme liefen die simulierten Routen nah — innerhalb weniger Ångström — an Zwischenstrukturen vorbei, die nicht als Eingabe bereitgestellt worden waren. Bei anspruchsvollen Fällen wie dem DNA-Reparaturenzym DNA-PKcs oder dem Coronavirus-Spike-Protein überlappten die grobkörnigen Pfade außerdem gut mit deutlich teureren atomaren Simulationen, einschließlich gezielter molekularer Dynamik und fortgeschrittener Enhanced-Sampling-Läufe.
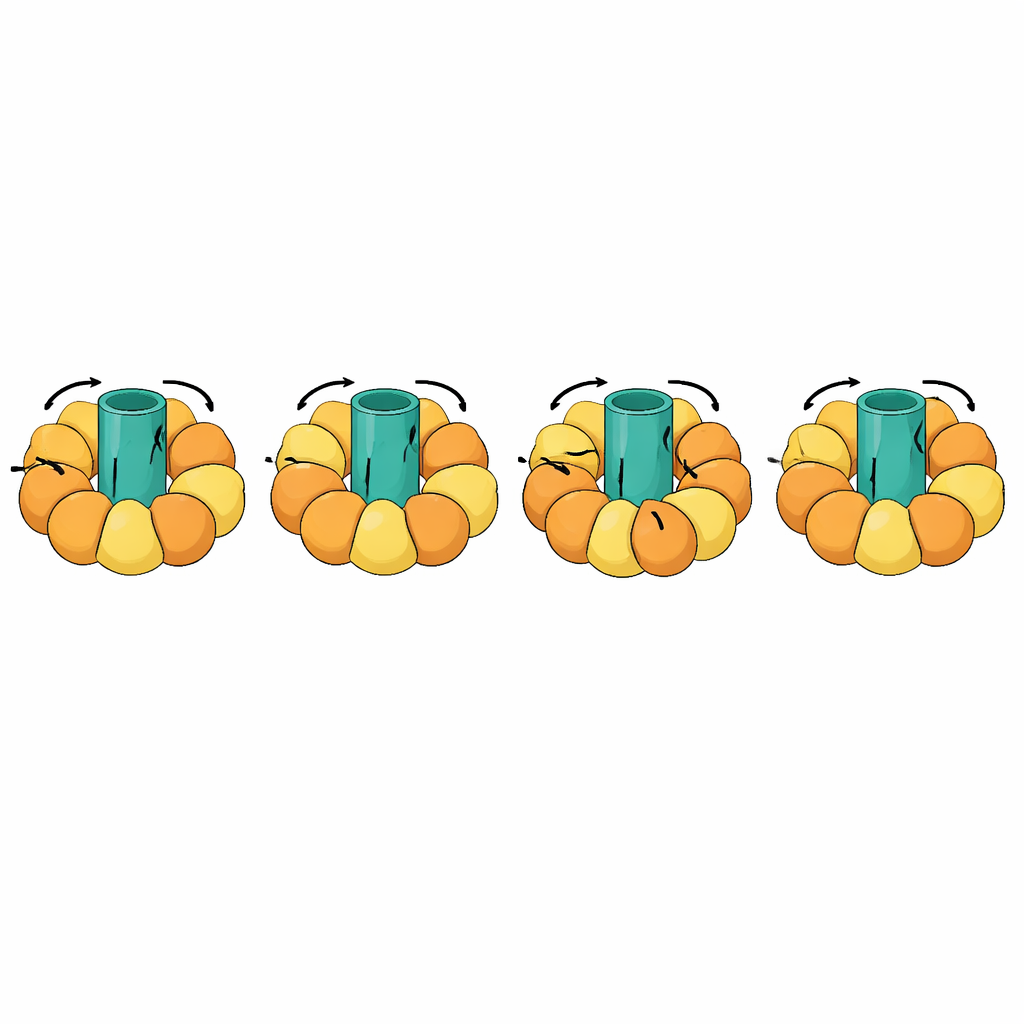
Riesenhafte molekulare Maschinen verfolgen
Eine der eindrucksvollsten Prüfungen betraf rotierende Maschinen wie ATP-Synthasen, die die Energiewährung der Zelle herstellen, indem sie einen rotierenden Rotor in der Membran mit Öffnungs- und Schließbewegungen in umgebenden Untereinheiten koppeln. Diese Übergänge sind außerordentlich komplex: Teile des Moleküls müssen starr bleiben und als Einheit rotieren, während andere sich in einem choreografierten Zyklus biegen. eBDIMS2 führt spezielle Behandlung für solche quasi-starren Teile und für unvollständige experimentelle Modelle mit fehlenden Segmenten ein, beides häufige Befunde in der Kryo-Elektronenmikroskopie. Mit diesen Funktionen kann es vollständige Rotationszyklen von ATP-Synthase und anderen massiven Komplexen wie molekularen Chaperonen, Rezeptoren und viralen Assemblies simulieren. Dabei vermeiden die erzeugten Zwischenstrukturen die starken Verzerrungen, die von einigen konkurrierenden Methoden produziert werden, und lassen sich zu atomistischen Modellen aufbereiten, die sich für Wirkstoffdesign-Berechnungen oder längere, detailliertere Simulationen eignen.
Was das für Biologie und Medizin bedeutet
Die Studie zeigt, dass eBDIMS2 zuverlässig die Hauptwege zwischen bekannten Proteinformen für Systeme skizzieren kann, die für traditionelle Simulationen unerreichbar waren. Es ersetzt keine detaillierten atomaren Filme und liefert keine präzisen Energien oder Zeiten, bietet jedoch eine schnelle, physikalisch fundierte Möglichkeit, zu kartieren, wie große molekulare Maschinen sich bewegen könnten, und zwar allein auf Basis eines Paars experimenteller Strukturen als Eingabe. Da Struktur-Datenbanken zunehmend mehrere Zustände großer Proteinassemblies enthalten, die mit Krebs, Infektionen und anderen Krankheiten verknüpft sind, bietet dieser Ansatz Forschern ein zugängliches Werkzeug, um die Verbindungen herzustellen, plausible Zwischenzustände vorzuschlagen und zu leiten, wo mit höherauflösenden Methoden oder gezieltem Wirkstoffdesign als Nächstes gesucht werden sollte.
Zitation: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Schlüsselwörter: Proteindynamik, molekulare Simulationen, cryo-EM, Konformationspfade, grobkörniges Modellieren