Clear Sky Science · de
Aktivierung von IRF3 in Kardiomyozyten beeinträchtigt die mitochondriale oxidative Funktion durch Hemmung von PGC-1α und treibt Herzinsuffizienz voran
Warum gestresste Herzen und erschöpfte Zellen wichtig sind
Herzinsuffizienz wird oft als ein „Verschleiß“ des Herzens beschrieben, doch unter der Oberfläche ist es auch eine Geschichte von chronischer Entzündung und erschöpften Kraftwerken in den Herzmuskelzellen. Diese Studie stellt eine auf den ersten Blick einfache, aber folgenreiche Frage: Gibt es einen einzelnen molekularen Schalter in Herz-Zellen, der schädliche Entzündung und versagende Energieproduktion verbindet — und wenn ja, kann das Umlegen dieses Schalters den Verlauf der Herzinsuffizienz verändern? Indem die Autorinnen und Autoren diesem Faden folgen, identifizieren sie einen Schlüsselakteur und zeigen, dass ein moderates Anheben des körpereigenen Energieprogramms das Versagen des Herzens bei Mäusen teilweise rückgängig machen kann.
Ein molekularer Schalter in kranken menschlichen Herzen
Die Forschenden konzentrierten sich auf ein Protein namens IRF3, das vor allem dafür bekannt ist, Zellen bei Virusinfektionen zu unterstützen. Sie untersuchten Gewebe von Menschen mit ischämischer Kardiomyopathie, einer häufigen Form der Herzinsuffizienz, die durch verminderten Blutfluss nach Herzinfarkten entsteht. In diesen versagenden Herzen war IRF3 nicht nur vorhanden — es war an bestimmten Stellen chemisch aktiviert, ein Hinweis darauf, dass es aktiv Genprogramme steuerte. Gleichzeitig war die Maschinerie, mit der Mitochondrien Brennstoff mittels oxidativer Phosphorylierung in Energie umwandeln, deutlich geschwächt. Ein ähnliches Muster zeigte sich in Mausmodellen von Herzinfarkt: Wenn eine Koronararterie verschlossen wurde, wurde IRF3 in Herzmuskelzellen stark aktiviert und IRF3-regulierte Gene wurden hochgefahren. Sogar Fragmente mitochondrialer DNA — freigesetzt von beschädigten Mitochondrien und als interne „Gefahr“-Signale wirkend — reichten aus, um IRF3 in isolierten Herz-Zellen zu aktivieren.
IRF3 ausschalten schützt das Herz
Um zu prüfen, ob die IRF3-Aktivität in Herzmuskelzellen die Erkrankung tatsächlich verschlimmert, erzeugte das Team Mäuse, bei denen IRF3 nur in Kardiomyozyten entfernt werden konnte, während Immun- und Stützzellen unberührt blieben. Nach Auslösung eines Herzinfarkts zeigten diese Mäuse trotz gleicher Anfangsverletzung eine bessere Pumpfunktion und weniger Narbenbildung als normale Mäuse. In kultivierten Herz-Zellen dämpfte das Abschalten von IRF3 entzündliche Gene, ohne verwandte Proteine zu stören. Zusammen sprechen diese Befunde dafür, dass IRF3 innerhalb der Herz-Zelle kein bloßer Beobachter ist: Es verstärkt Entzündung und strukturelle Schäden nach Ischämie und fördert so den Übergang zur Herzinsuffizienz.
Wenn IRF3 dauerhaft „ein“ steht, bricht das Brennstoffsystem zusammen
Die Autorinnen und Autoren kehrten das Experiment um: Sie erzeugten Mäuse, bei denen IRF3 in Kardiomyozyten durch einen genetischen „Phosphomimetik“-Trick dauerhaft aktiv gehalten werden konnte. Selbst ohne äußeren Auslöser entwickelten diese Mäuse rasch schwere Herzfunktionsstörungen, hohe Spiegel entzündlicher Botenstoffe im Blut und Anzeichen zellulärer Verletzung. Eine detaillierte Analyse ihres Herzgewebes zeigte, dass chronisch aktives IRF3 einen zentralen Energieregulator namens PGC-1α unterdrückt. Dieses Molekül fördert normalerweise gesunde Mitochondrien, effiziente Fettverbrennung und ausgewogene zelluläre Energie. Mit gedämpftem PGC-1α fielen zahlreiche mitochondriale Proteine ab, die Elektronentransportkette schwächelte, und die Treibstoffnutzung des Herzens verschob sich: Carnitin und verwandte Verbindungen für die Fettverbrennung sanken, die Nutzung von Ketonen war eingeschränkt und der Glukosestoffwechsel wurde verzerrt. Sogar das Verhältnis von NAD⁺ zu NADH — ein wichtiger Indikator des zellulären Redoxgleichgewichts — verschob sich in die falsche Richtung.
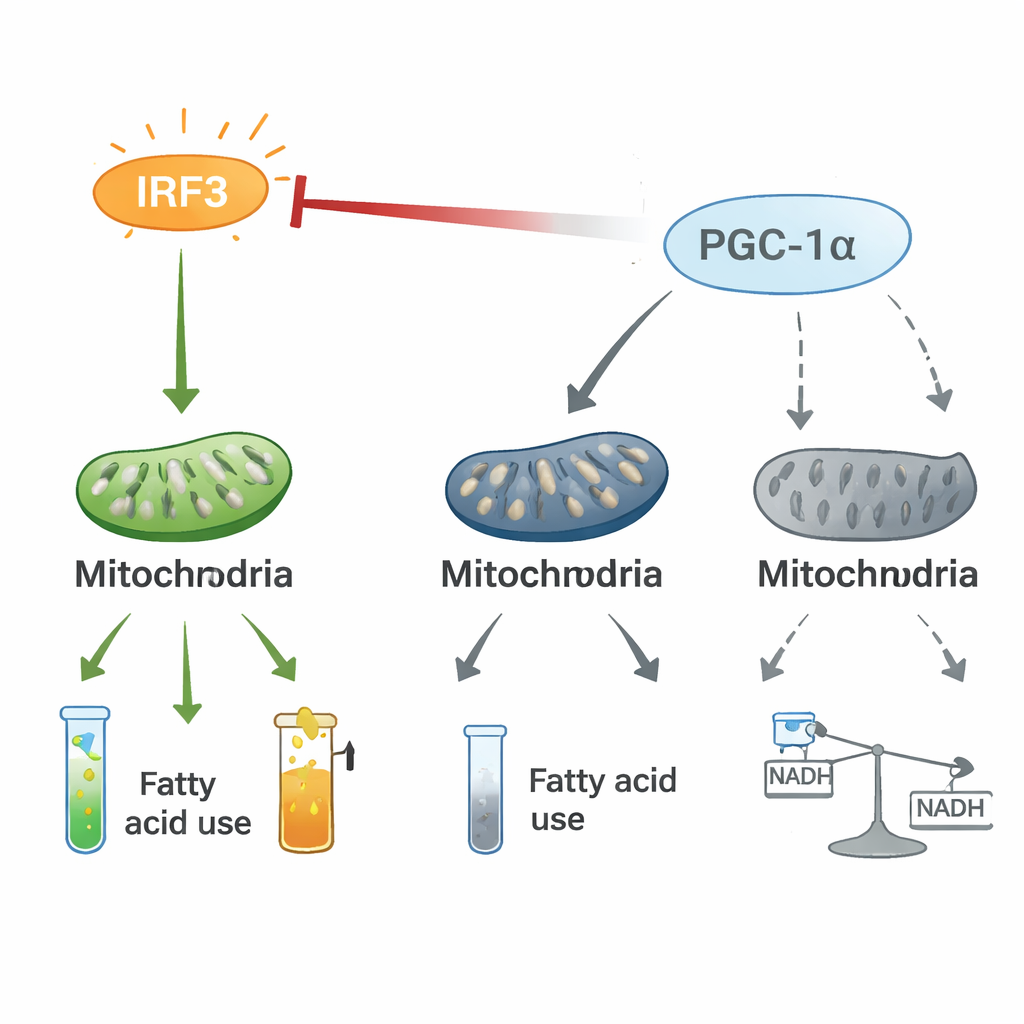
Ein Tauziehen zwischen Entzündung und Energiesteuerung
Mechanistische Experimente zeigten, dass IRF3 und PGC-1α eine zweiseitige Regulationsachse bilden. In Herz-Zellen geht aktiviertes IRF3 eine physische Wechselwirkung mit PGC-1α ein und schwächt dessen Fähigkeit, Gene der Fettverbrennung zu aktivieren. Das Herunterregeln von IRF3 erhöht PGC-1α-Spiegel und -Aktivität, während eine Erhöhung von PGC-1α IRF3-getriebene entzündliche Gene dämpft und mitochondriale Marker wiederherstellt — selbst unter Stressbedingungen wie Sauerstoffmangel oder bakteriellen Toxinen. Stabil markiertes Isotopen-Tracking zeigte, dass IRF3-Aktivierung Kohlenstoff aus der normalen Energieproduktion über den Zitratzyklus in alternative Wege wie die Pentosephosphatweg umlenkt und den reibungslosen Fluss von Metaboliten stört. Dieses Tauziehen zwischen einem pro-inflammatorischen Schalter (IRF3) und einem Energie-Co-Piloten (PGC-1α) scheint den Stoffwechsel des Herzens so umzubauen, dass Entzündung und Energieverlust begünstigt werden.
Die Batterien des Herzens behutsam wieder aufladen
Schließlich fragten die Forschenden, ob ein leichtes Anheben von PGC-1α die Schäden durch IRF3 ausgleichen könnte. Sie verwendeten einen herzgerichteten Gentherapie-Vektor, um PGC-1α moderat — aber nicht übermäßig — in denselben Mäusen mit hyperaktivem IRF3 zu erhöhen. Dieser moderate Schub verbesserte die Pumpfunktion, erhöhte mitochondriale Proteine, förderte Gene für Fettverbrennung und NAD-Stoffwechsel und reduzierte entzündliche sowie fibrotische Genaktivität. In Zellversuchen stellte die Ko-Expression von PGC-1α mit aktivem IRF3 ein gesünderes NAD⁺/NADH-Verhältnis wieder her und verschob die Treibstoffnutzung zurück in Richtung Fettsäuren. Für Laien bedeutet das: Ein behutsames Wiederaufladen des „Batteriemanagementsystems“ des Herzens kann die schädlichen Effekte eines chronisch eingeschalteten entzündlichen Schalters teilweise ausgleichen.
Was das für die künftige Herzinsuffizienzbehandlung bedeutet
Diese Arbeit positioniert IRF3 als zentrale Verbindung zwischen Entzündung und Energieversagen in Herzmuskelzellen. Anstatt Entzündung und Stoffwechsel in der Herzinsuffizienz als getrennte Probleme zu behandeln, legt die Studie nahe, dass sie durch eine IRF3–PGC-1α-Achse miteinander verknüpft sind. Obwohl diese Befunde in Mäusen und Zellmodellen gewonnen wurden, eröffnen sie die Möglichkeit, dass künftige Therapien entweder die IRF3-Aktivität drosseln oder PGC-1α und die mitochondriale Funktion stärken könnten, um Herzinsuffizienz nach einem Herzinfarkt zu verlangsamen oder zu verhindern. Kurz gesagt: Das Beruhigen eines überaktiven zellulären Alarmsystems und die Unterstützung der Energiefabriken des Herzens könnten zusammen eine wirksame Strategie sein, um geschwächte Herzen länger kräftig schlagen zu lassen.
Zitation: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Schlüsselwörter: Herzinsuffizienz, Entzündung, Mitochondrien, Kardiomyozyten, PGC-1α