Clear Sky Science · de
GCN5 treibt das Fortschreiten von MASLD über den LXRα/SREBP1c-Signalweg–vermittelte de-novo-Lipogenese voran
Warum diese Leberstudie wichtig ist
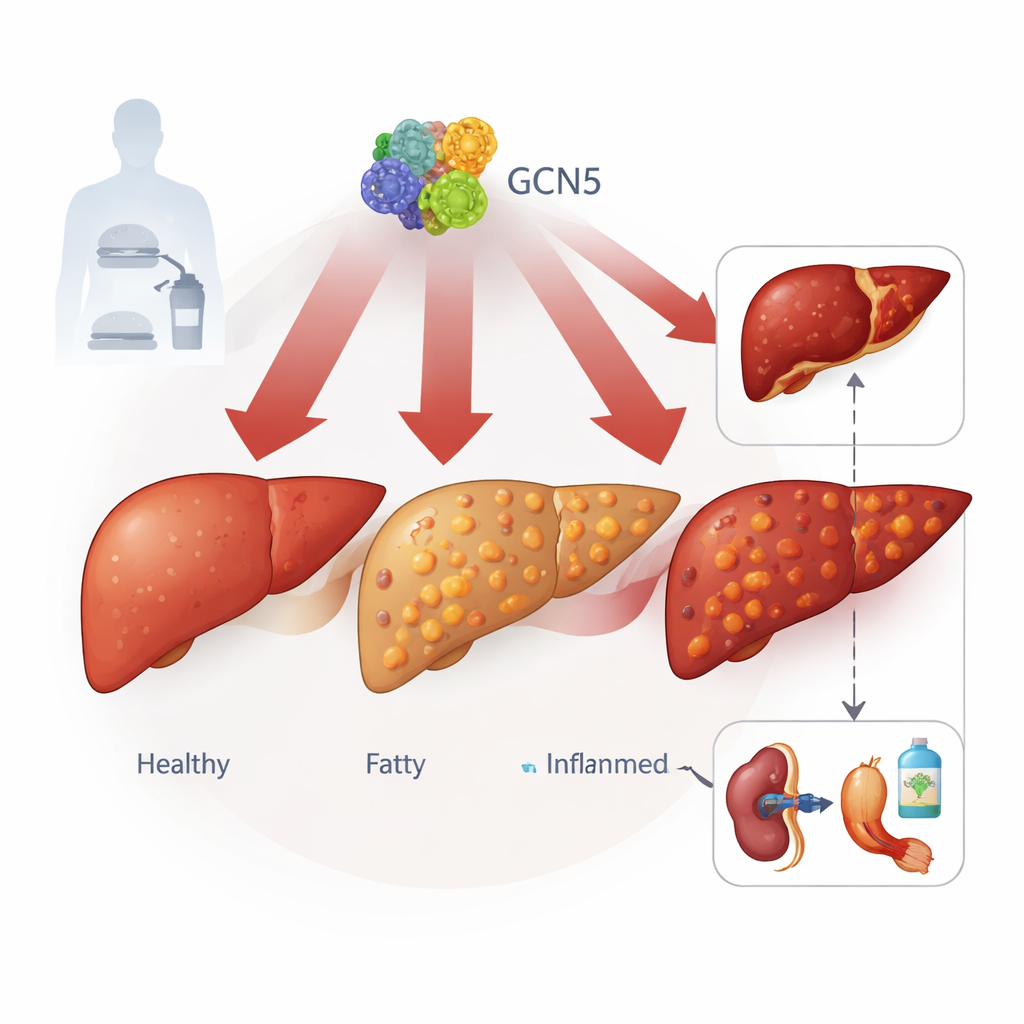
Die metabolisch bedingte, steatotische Lebererkrankung (MASLD), früher als nichtalkoholische Fettleberkrankheit bezeichnet, betrifft heute weltweit etwa jeden vierten Menschen. Sie verläuft oft unbemerkt, kann aber zu Vernarbung, Leberkrebs und schweren Stoffwechselstörungen fortschreiten. Diese Studie deckt einen molekularen "Lautstärkeregler" auf, der die schädliche Fettansammlung in der Leber fördert, und zeigt, wie dessen Abschwächung die Leber schützen und gleichzeitig bestehende kardiovaskuläre Therapiestrategien sicherer machen könnte.
Ein versteckter Schalter in Leberzellen
Die Autor:innen konzentrieren sich auf ein Protein namens GCN5, das vor allem als Regulator der DNA-Packung bekannt ist. Anhand von Lebergewebeproben von mehr als 100 Menschen – von gesunden bis zu schwer betroffenen MASLD-Fällen – sowie mehreren Mausmodellen einer diätinduzierten Fettleber fanden sie, dass die GCN5-Spiegel in Leberzellen mit Fortschreiten der Erkrankung stetig ansteigen. Ein eng verwandtes Protein, PCAF, zeigte dieses Muster nicht, was darauf hindeutet, dass GCN5 eine besondere Rolle spielt. Hohe GCN5-Werte korrelierten mit mehr Leberfett, höheren Blutfettwerten und stärkeren Zeichen von Leberschädigung und verknüpfen diesen molekularen Schalter mit der klinischen Schwere der Erkrankung.
GCN5 hoch- oder runterregeln bei Tieren
Um Ursache und Wirkung zu prüfen, erhöhten bzw. entfernten die Forschenden GCN5 gezielt in Leberzellen von Mäusen. Wurde GCN5 hochreguliert, entwickelten Mäuse unter einer fettreichen Diät schwerere, fettreichere Lebern, höhere Blutlipide und stärkere Leberschädigungen, obwohl sie nicht mehr oder deutlich schwerer aßen. In kultivierten Leberzellen verhielt es sich ähnlich: Mehr GCN5 führte zu größeren und zahlreicheren Fetttröpfchen. Im Gegensatz dazu waren Mäuse, bei denen GCN5 nur in den Leberzellen fehlte, deutlich geschützt. In mehreren Diätmodellen, die menschliches MASLD und seine entzündlichere Form nachahmen, akkumulierten diese Tiere weniger Leberfett, zeigten geringere Blutlipide und Leberenzyme und entwickelten weniger Entzündungen und Vernarbung.
Wie GCN5 die Leber zur Fettherstellung antreibt
Bei einem genaueren Blick auf den Stoffwechsel bestimmten die Forschenden zahlreiche Fettsäuren und deren Bausteine in der Leber. Der Verlust von GCN5 verringerte vor allem diejenigen Fette, die die Leber neu aufbaut – ein Prozess, der als de-novo-Lipogenese bezeichnet wird –, während diätbedingt zugeführte mehrfach ungesättigte Fettsäuren weitgehend unverändert blieben. Untersuchungen zur Genexpression und Isotopenverfolgung zeigten, dass GCN5 oberhalb eines zentralen Fettbildungsregulators namens SREBP1c angesiedelt ist. Wenn GCN5 aktiv war, wurden Gene, die Fettsäuren aufbauen und modifizieren, eingeschaltet und die interne Fettherstellungsrate der Leber stieg. Das Entfernen oder Blockieren von GCN5 drosselte dieses Programm und reduzierte die Flussmenge von Kohlenstoff aus Zucker in neu gebildetes Leberfett.
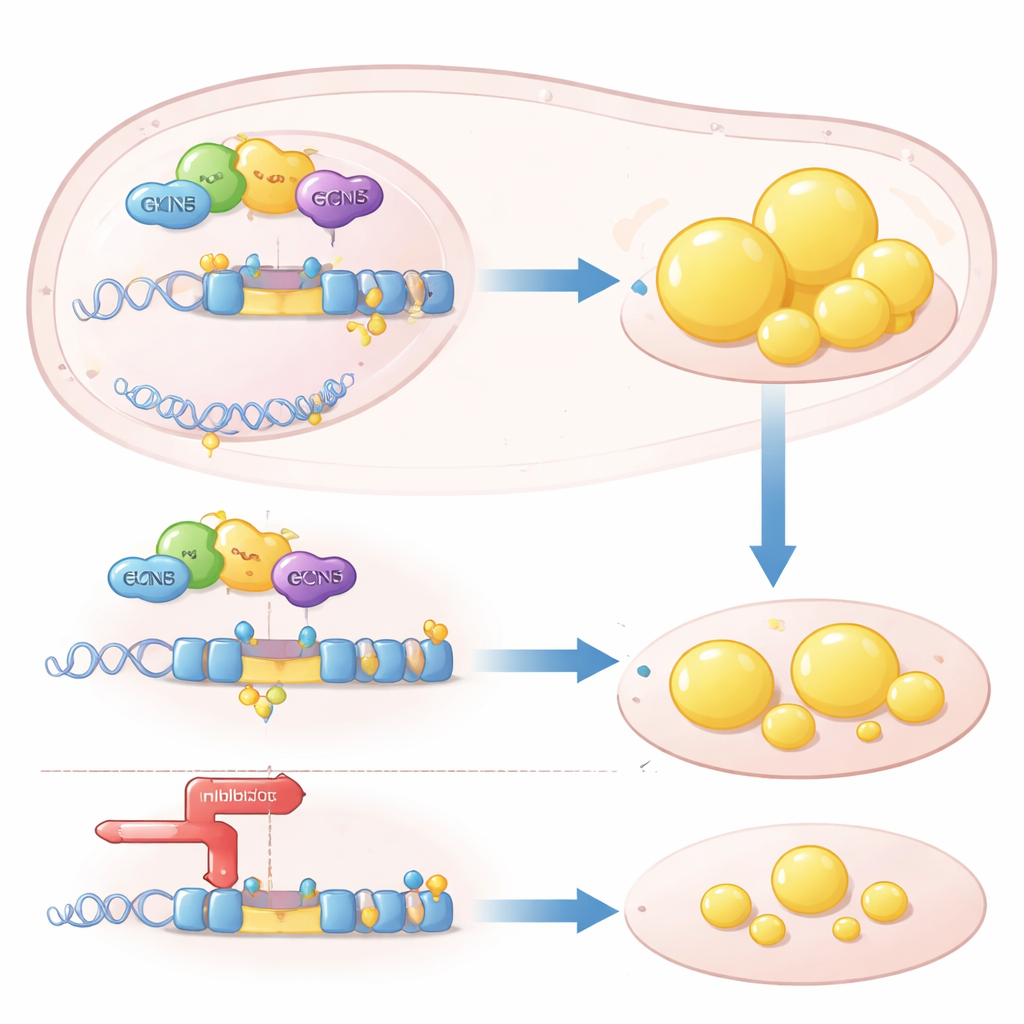
Ein epigenetischer Hebel an einem Schlüsselfettgen
Mechanistisch wirkt GCN5 als ein epigenetischer Hebel: Es verändert chemisch Histonproteine an der DNA, sodass bestimmte Gene leichter abgelesen werden können. Die Autor:innen zeigten, dass GCN5 zusammen mit einem Kernrezeptor namens LXRα, der cholesterinähnliche Moleküle erkennt, an die Kontrollregion des SREBP1c-Gens rekrutiert wird. Dort setzt GCN5 Acetylgruppen an Histon H3, lockert das lokale Chromatin und verstärkt die Transkription von SREBP1c. Dieser Effekt war hochselektiv: GCN5 verstärkte LXRαs Fähigkeit, SREBP1c zu aktivieren, nicht jedoch ein anderes LXR-Ziel, ABCA1, das beim Cholesterinentzug aus Geweben hilft. Ohne GCN5 konnte LXRα den SREBP1c-Promotor nicht mehr effizient ansprechen, und das nachgeschaltete Fettsyntheseprogramm stagnierte.
Ein Arzneistoffkandidat und eine vielversprechende Kombination
Die Forschenden testeten anschließend CPTH2, einen kleinmolekularen Hemmstoff von GCN5, der sich in der Leber anreichert. In Mausmodellen, die bereits fettreiche Diäten erhielten, verringerte CPTH2 Lebergröße, Fettgehalt und Schadstoffmarker ohne offensichtliche Toxizität oder Veränderungen der Nahrungsaufnahme. In kultivierten menschlichen und Maus-Leberzellen senkte CPTH2 Fetttröpfchen und Triglyceride nur, wenn GCN5 vorhanden war, was die Spezifität seiner Wirkung bestätigte. Wichtig ist, dass CPTH2 in Zellen und Mäusen, die mit LXR-aktivierenden Verbindungen behandelt wurden (die darauf abzielen, Cholesterin zu entfernen und Atherosklerose zu bekämpfen), selektiv den unerwünschten Anstieg der SREBP1c-getriebenen Fettsynthese blockierte, während Gene, die den Rücktransport von Cholesterin fördern, erhalten blieben. In Kombination mit einem LXR-Agonisten bei hochfetthaltiger Fütterung senkte CPTH2 darüber hinaus schädliche Blutlipide und Lebercholesterin und verhinderte zusätzliche Fettansammlung in der Leber.
Was das für Patient:innen bedeutet
Die Studie positioniert GCN5 als zentralen Treiber der Leberfettansammlung bei MASLD, indem sie diätetische und hormonelle Signale mit dem SREBP1c-Fettherstellungsschalter verknüpft. Da GCN5 offenbar für den vorteilhaften, cholesterinentziehenden Arm der LXR-Signalgebung entbehrlich ist, könnten GCN5-hemmende Wirkstoffe – wie CPTH2 oder weiterentwickelte Nachfolger – Leberfett und Entzündungen dämpfen und gleichzeitig herzschützende Therapien ermöglichen. Für Menschen mit Risiko sowohl für Fettlebererkrankung als auch Herz-Kreislauf-Erkrankungen könnte das Ansteuern dieses epigenetischen Schalters eines Tages einen Weg bieten, die Leber zu schützen, ohne die Vorteile einer verbesserten Cholesterinregulation zu opfern.
Zitation: Xiao, HT., Song, P., Jin, J. et al. GCN5 drives MASLD progression through LXRα/SREBP1c signaling pathway–mediated de novo lipogenesis. Nat Commun 17, 2821 (2026). https://doi.org/10.1038/s41467-026-69736-y
Schlüsselwörter: Fettlebererkrankung, Epigenetik, Lipidstoffwechsel, Leberstoffwechsel, Kernrezeptoren