Clear Sky Science · de
Erweiterung der Reichweite von Graph-Neuronalen Netzen mit globalen Codierungen
Warum Verbindungen über weite Entfernungen in Molekülen wichtig sind
Von neuen Arzneimitteln bis zu besseren Batterien beruhen viele aktuelle Durchbrüche auf Computermodellen, die vorhersagen können, wie Tausende von Atomen aufeinander einwirken. Eine verbreitete Klasse von KI-Modellen, sogenannte graph neuronale Netze, ist dafür zu einem Standardwerkzeug geworden. Diese Modelle haben jedoch eine Schwäche: Sie richten ihren Blick hauptsächlich auf unmittelbare Nachbarn, obwohl entfernte Atome durch elektrische und quantenmechanische Kräfte starken Einfluss aufeinander ausüben können. Dieser Artikel stellt RANGE vor, eine Methode, die diesen neuronalen Netzen eine Art globale Sicht verleiht, sodass sie Langstreckeneffekte „wahrnehmen“ und vorhersagen können, ohne dabei unangemessen langsam oder speicherintensiv zu werden.
Wie aktuelle KI nur die Nachbarschaft sieht
Graph neuronale Netze behandeln ein Molekül oder Material als ein Netz von Knoten (Atome), die über Kanten (Beziehungen) verbunden sind. In jedem Schritt aktualisiert jeder Knoten seinen Zustand, indem er nur mit seinen nahegelegenen Nachbarn innerhalb einer festen Distanz kommuniziert. Durch viele Wiederholungen verbreiten sich die Informationen langsam, doch diese Strategie hat zwei große Nachteile. Erstens können Botschaften beim Durchlaufen vieler Zwischenstationen verwischen — ein Problem, das als Oversmoothing bekannt ist. Zweitens können schmale Pfade im Graphen die Informationsmenge drosseln, was zu Oversquashing führt. Beide Probleme werden gravierend, wenn man Kräfte erfassen möchte, die über viele Ångström wirken, wie Elektrostatik und Dispersionskräfte in großen Molekülen oder Kristallen. Einfach die Interaktionsreichweite zu vergrößern oder mehr Schichten übereinander zu stapeln macht die Modelle teuer und löst die Engpässe nicht vollständig.
Hinzufügen virtueller Hubs für globale Kommunikation
RANGE (Relaying Attention Nodes for Global Encoding) verändert dieses Bild, indem es eine kleine Anzahl virtueller „Masterknoten“ einführt, die keinem realen Atom entsprechen. Stattdessen fungieren sie als globale Hubs. Nach einem gewöhnlichen Message-Passing-Schritt zwischen benachbarten Atomen werden Informationen aller Atome durch einen Aufmerksamkeitsmechanismus in diese Hubs eingesammelt: Jeder Masterknoten lernt, auf welche Teile des Systems er sich konzentrieren sollte. Diese Aggregation erzeugt grob zusammengefasste Darstellungen des Zustands des Moleküls. In einem zweiten Broadcast-Schritt werden diese Zusammenfassungen zurück an jedes Atom gesendet — wiederum mit Attention, sodass jedes Atom entscheiden kann, wie viel es von jedem Masterknoten aufnehmen möchte, während es durch Self-Loops seine lokale Erinnerung bewahrt. Da jedes Atom direkt mit jedem Masterknoten verbunden ist, kann Langstreckenkommunikation in einem einzigen Schritt stattfinden und den Graphen in ein Small-World-Netz verwandeln, in dem weit auseinanderliegende Regionen schnell und effizient aufeinander einwirken können. 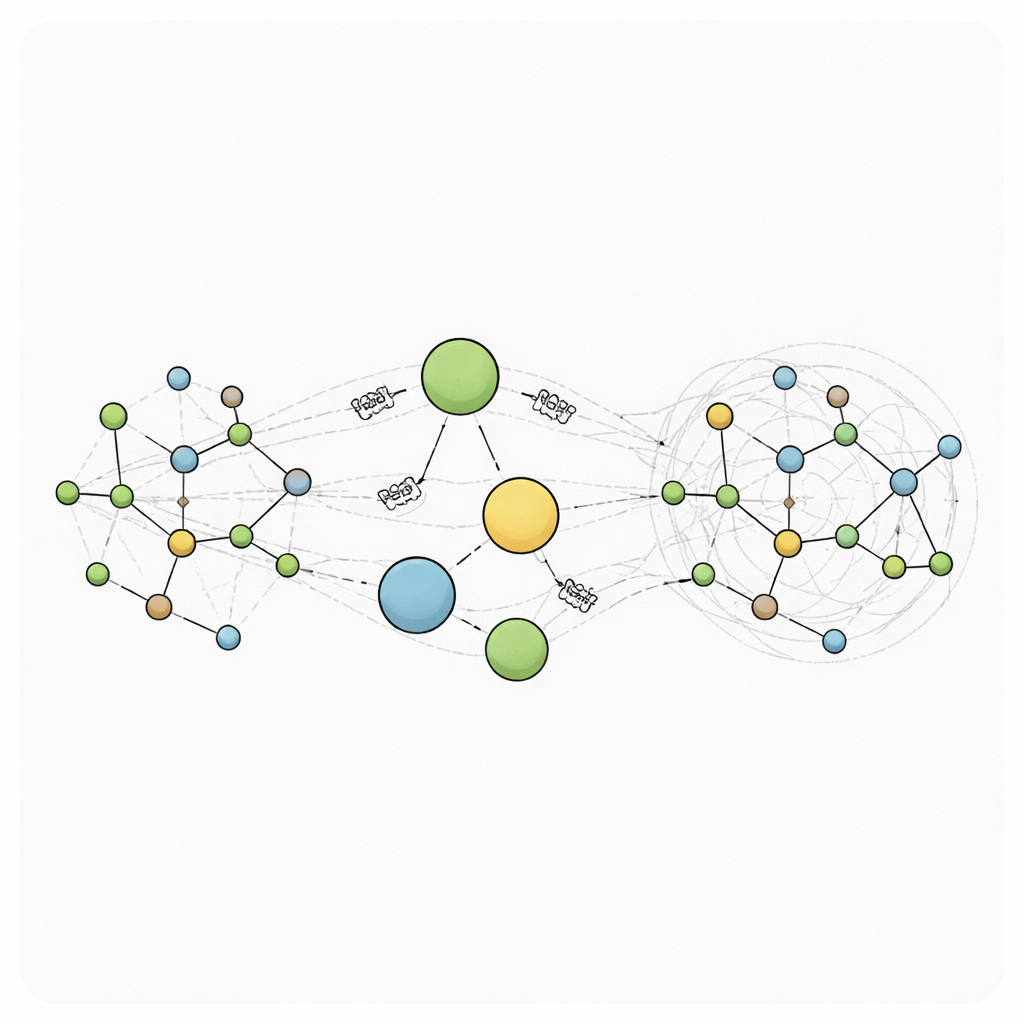
Langstreckenkräfte sehen, die andere übersehen
Die Forscher prüften RANGE, indem sie es an mehrere hochmoderne Modelle für molekulare Kraftfelder anbanden und mit ihren ursprünglichen, rein lokalen Versionen verglichen. Sie wählten herausfordernde Systeme, in denen Langreichweiten-Effekte bekanntlich entscheidend sind: einen Salzkristall mit einem zusätzlichen Natrium-Ion als Dopant, ein Golddimer, das sich einer dotierten Oxidoberfläche nähert, und Paare organischer Moleküle, die in variierenden Abständen interagieren. Standardmodelle verpassten weitgehend, wie entfernte Ladungsumverteilungen oder versteckte Dopanten die Energielandschaft veränderten; ihre Vorhersagen verschoben sich kaum, wenn sich die Umgebung auf lange Distanz änderte. Im Gegensatz dazu erfassten die mit RANGE erweiterten Modelle korrekt die unterschiedlichen Energieprofile und konnten auf größere Abstände extrapolieren, als sie im Training gesehen hatten, mit bis zu viermal geringeren Fehlern bei schwierigen geladenen Dimeren.
Genauigkeit ohne den Computer zu überlasten
Wesentlich ist, dass RANGE diese verbesserte Übersicht liefert, ohne die hohen Rechenkosten anderer globaler Ansätze. Techniken, die sich physikalischer Methoden bedienen, wie Ewald-Summen oder Fourieranpassungen, erfordern Operationen, die ungefähr mit dem Quadrat der Atomanzahl wachsen oder von großen Gittern abhängen, was sie bei großen Systemen und wiederholten Simulationen schwerfällig macht. RANGE skaliert hingegen per Design linear mit der Systemgröße: Jeder Masterknoten verbindet sich mit allen Atomen, aber die Anzahl der Masterknoten wächst moderat und wird durch ein Regularisierungsschema gesteuert, das Redundanz verhindert. Benchmarks auf umfangreicheren Datensätzen zeigen, dass RANGE konstant Fehler in den vorhergesagten Kräften reduziert, selbst wenn die zugrunde liegenden Modelle kurze Interaktionsschnittstellen verwenden, und das bei nur moderatem Mehraufwand an Laufzeit und Speicher. Das Team führte außerdem Molekulardynamik-Simulationen über Dutzende Nanosekunden an komplexen Molekülen durch und fand, dass RANGE-basierte Kraftfelder stabil blieben und realistische Konformationen und Zustände erkundeten. 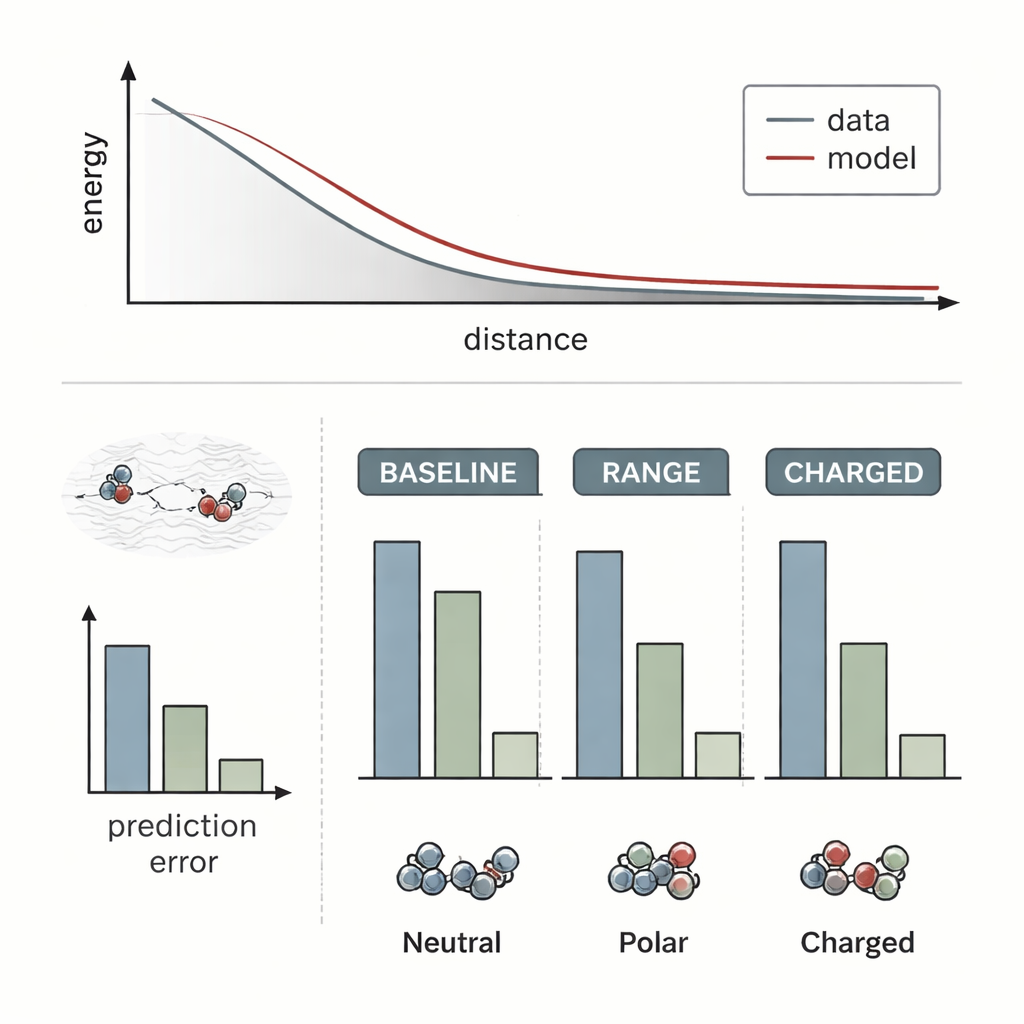
Ein klareres Gesamtbild molekularer Welten
Für Nicht-Spezialisten ist die Kernbotschaft: RANGE verleiht bestehenden graphbasierten KI-Modellen eine neue Möglichkeit, global zu denken, ohne das lokale Arbeiten aufzugeben. Durch intelligente virtuelle Hubs und auf Attention basierenden Informationsfluss überwindet es die üblichen Engpässe, die neuronalen Netzen das Erfassen langreichweitiger, viele-Körper-Effekte in Molekülen und Materialien erschweren. Das bedeutet verlässlichere Vorhersagen für Systeme, in denen weit auseinanderliegende Bereiche sich subtil beeinflussen — von flexiblen Wirkstoffmolekülen bis zu ausgedehnten Nanostrukturen — ohne eine unvertretbar hohe Rechenrechnung. Wenn diese Methoden auf immer größere und komplexere Umgebungen angewendet werden, versprechen sie KI-Werkzeuge, die das wahre, langreichweitige Gefüge physikalischer Wechselwirkungen treuer abbilden können.
Zitation: Caruso, A., Venturin, J., Giambagli, L. et al. Extending the range of graph neural networks with global encodings. Nat Commun 17, 1855 (2026). https://doi.org/10.1038/s41467-026-69715-3
Schlüsselwörter: graph neuronale netze, wechselwirkungen über große entfernungen, molekulare Simulationen, maschinell gelernte Kraftfelder, Aufmerksamkeitsmechanismen