Clear Sky Science · de
Kombination von PARP- und KRASG12D-Inhibitoren erhöht die therapeutische Wirksamkeit durch Ausnutzung von Verwundbarkeiten bei PDAC
Warum diese Studie wichtig ist
Pankreaskrebs gehört zu den tödlichsten häufigen Krebsarten, vor allem weil er meist spät entdeckt wird und Standardbehandlungen oft wenig anhaben können. Viele dieser Tumoren werden durch einen spezifischen genetischen Defekt namens KRASG12D angetrieben; ein neues experimentelles Medikament wirkt gegen diesen Defekt, stößt aber schnell auf Resistenz. Die Studie stellt eine pragmatische Frage mit realen Folgen: Kann man das KRAS-blockierende Medikament mit einem zweiten Wirkstoff kombinieren, um eine kurzlebige Reaktion in einen tieferen, länger anhaltenden Angriff auf den Krebs zu verwandeln?
Ein hartnäckiger Krebs mit einer gemeinsamen Schwachstelle
Die meisten Pankreasduktaladenokarzinome tragen Mutationen im KRAS-Gen, das wie ein klemmendes Gaspedal für Zellwachstum wirkt. Unter diesen ist die KRASG12D-Variante sowohl die häufigste als auch am stärksten mit schlechter Überlebensprognose verknüpft. Die Forschenden bestätigten zunächst mithilfe großer Krebsdatenbanken, dass Patientinnen und Patienten mit Tumoren, die diese Mutation tragen, tendenziell schlechter abschneiden als solche mit anderen KRAS-Veränderungen oder ohne Mutation. Außerdem fiel auf, dass KRASG12D-Tumoren eine hohe Aktivität in Genen zeigen, die beschädigte DNA reparieren — ein Hinweis darauf, dass diese Krebszellen auf leistungsfähige DNA-Reparaturmechanismen angewiesen sein könnten, um dem ständigen Schaden durch schnelles Wachstum zu trotzen.
Eine Stärke in eine Schwäche verwandeln
Das Team untersuchte ein hochselektives KRASG12D-blockierendes Medikament namens MRTX1133 in im Labor gezüchteten Pankreastumorzellen. Behandelten sie KRASG12D-mutierte Zellen mit diesem Wirkstoff und setzten sie anschließend DNA-schädigender Strahlung aus, hatten die Zellen Probleme, ihre gebrochene DNA zu reparieren. Molekulare Tests zeigten den Grund: MRTX1133 senkte die Mengen wichtiger Reparaturproteine, darunter BRCA1 und RAD51, die normalerweise bei der Behebung gefährlicher Doppelstrangbrüche in der DNA helfen. Spezialisierte Reporter-Assays bestätigten, dass die Zellen „homologe Rekombinationsdefizient“ geworden waren — schlicht gesagt, sie verloren eines ihrer genauesten DNA-Reparatursysteme.
Kombination zweier zielgerichteter Medikamente für einen stärkeren Schlag
Der Verlust dieses Reparaturwegs ist genau die Art von Defekt, die Zellen gegenüber einer anderen Wirkstoffklasse — den PARP-Inhibitoren, die bereits bei bestimmten Brust- und Eierstockkrebserkrankungen eingesetzt werden — verwundbar macht. Die Forschenden kombinierten daher MRTX1133 mit dem PARP-Inhibitor Olaparib in KRASG12D-mutierten Pankreaskrebszellen und in Mausmodellen. In mehreren Zelllinien wirkten die beiden Medikamente zusammen deutlich besser als einzeln, töteten mehr Krebszellen und reduzierten ihre Fähigkeit, neue Kolonien zu bilden, stark. Bei Mäusen mit menschlichen oder murinen Pankreastumoren mit KRASG12D schrumpfte die Kombination die Tumoren tiefer und nachhaltiger als die Einzelsubstanzen und löste unter dem Mikroskop mehr DNA-Schäden und Krebszellsterben aus, während normale Zellen verschont blieben.
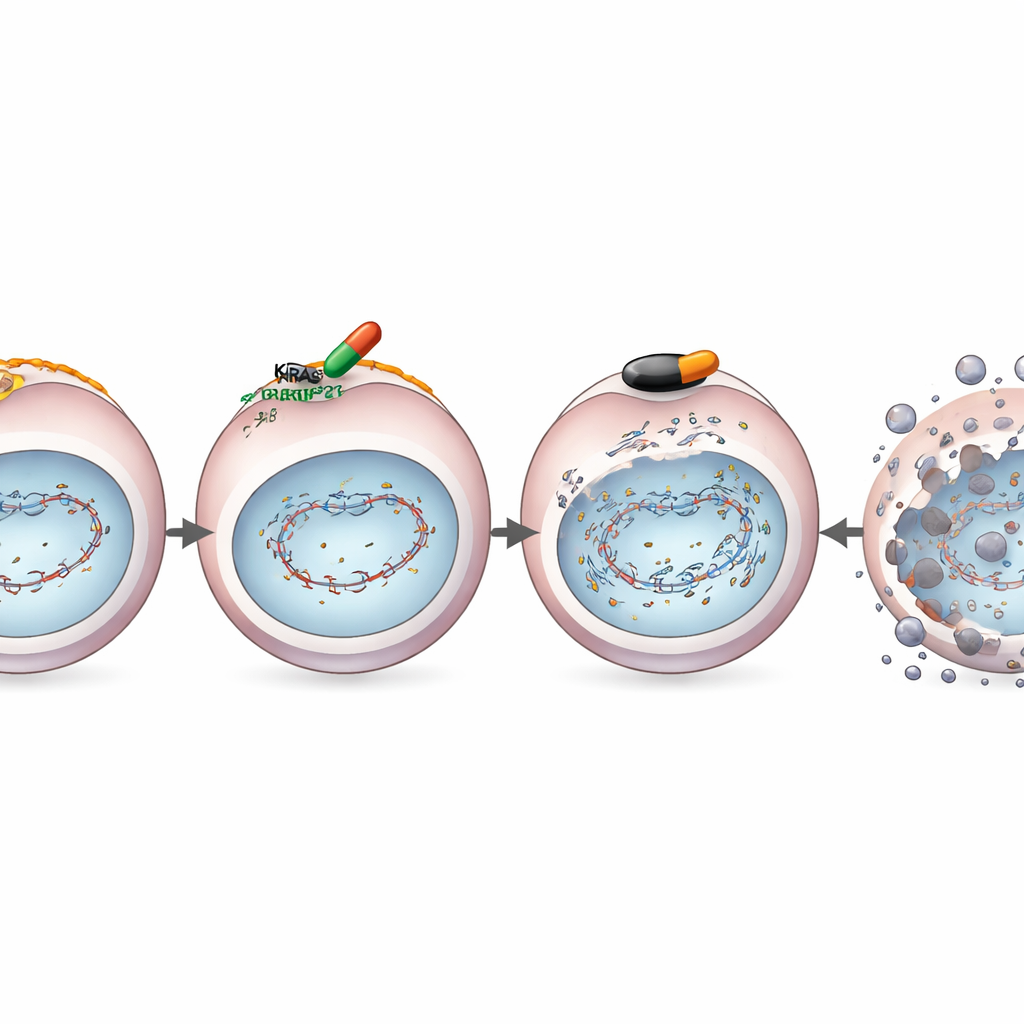
Wirksam auch wenn Resistenz auftritt
Zielgerichtete Medikamente wie MRTX1133 versagen oft, weil Tumoren ihre Wachstumswege umverdrahten und die Signalübertragung über alternative Routen wiederherstellen. Das Team erzeugte gezielt Krebszelllinien, die gegenüber den wachstumshemmenden Effekten von MRTX1133 resistent geworden waren. Auffallend war: Selbst in diesen resistenten Zellen drosselte das Medikament weiterhin BRCA1, RAD51 und verwandte Reparaturproteine und hielt die Schwäche in der DNA-Reparatur aufrecht. Folglich zeigte die Kombination aus MRTX1133 und Olaparib weiterhin eine starke, kooperative Zellabtötung in Kulturversuchen und in Mäusen mit resistenten Tumoren. Das legt nahe, dass die Kombination eine grundlegende Verwundbarkeit angreift, die selbst nach dem Einsetzen klassischer Resistenzwege bestehen bleibt.
Das Immunsystem wecken
Über die direkte Schädigung von Tumorzellen hinaus veränderte die Kombinationsbehandlung auch das Tumormikroumfeld. Mittels Einzelzell-RNA-Sequenzierung und Durchflusszytometrie in immunkompetenten Mäusen fanden die Forschenden, dass die Kombinationstherapie mehr krebsbekämpfende CD8- und helfende CD4-T-Zellen in die Tumoren zog und diese in einen aggressiveren „Effektor“-Zustand brachte, während Anzeichen von T-Zell-Erschöpfung abnahmen. Wurden CD8-T-Zellen experimentell entfernt, verringerte sich der Nutzen der Wirkstoffkombination, was zeigt, dass die Immunantwort ein wichtiger Teil des Gesamteffekts ist. Anders gesagt: Die Strategie schwächt den Tumor nicht nur von innen durch Beeinträchtigung der DNA-Reparatur, sie lädt auch das Immunsystem ein, sich dem Kampf anzuschließen.
Was das für Patientinnen und Patienten bedeuten könnte
Auch wenn der hier untersuchte spezifische KRASG12D-Wirkstoff klinisch nicht weiterverfolgt wird, liefert die Studie eine klare Botschaft: Die selektive Blockade von KRASG12D kann eine spezifische Schwäche in der DNA-Reparatur erzeugen, die Pankreastumoren besonders empfindlich gegenüber PARP-Inhibitoren macht — und das gilt selbst nachdem Resistenzen gegen den KRAS-Wirkstoff entstehen. Zukünftige KRASG12D‑gerichtete Präparate könnten mit PARP-Inhibitoren und möglicherweise mit Immuntherapien kombiniert werden, um eine einst „nicht ansprechbare“ Mutation in eine gezielte Behandlungsoption für den großen Anteil der Pankreaskrebspatientinnen und -patienten zu verwandeln, deren Tumoren diese genetische Veränderung tragen.
Zitation: Xu, X., Chen, X., Xu, R. et al. Combination of PARP and KRASG12D inhibitors enhances therapeutic efficacy by exploiting vulnerabilities in PDAC. Nat Commun 17, 3118 (2026). https://doi.org/10.1038/s41467-026-69695-4
Schlüsselwörter: Pankreaskrebs, KRASG12D, PARP-Inhibitor, DNA-Reparatur, Kombinationstherapie