Clear Sky Science · de
XL-MSDigger: eine tief lernende, vielseitige Lösung für Crosslinking-Massenspektrometrie
Sehen, wie Proteine zusammenhalten
Jeder Prozess in unseren Körpern hängt davon ab, dass Proteine nicht nur die richtige Faltung einnehmen, sondern auch die richtigen Partner finden. Diese molekularen Beziehungen in Aktion zu beobachten, ist jedoch notorisch schwierig. Diese Studie stellt XL-MSDigger vor, eine Softwareplattform, die moderne künstliche Intelligenz nutzt, um aus einer lauten experimentellen Methode namens Crosslinking-Massenspektrometrie deutlich klarere Signale zu extrahieren und so Wissenschaftlern zu helfen, die Anordnung von Proteinen und ihre Wechselwirkungen innerhalb von Zellen zu kartieren.
Entwirren einer überfüllten molekularen Welt
Um zu verstehen, wie Proteine aufgebaut sind und wie sie verbunden sind, verwenden Forscher häufig Crosslinking-Massenspektrometrie. Bei diesem Ansatz verbinden kleine chemische „Brücken“ nahe liegende Bereiche von Proteinen miteinander. Die verbundenen Stücke werden dann in Fragmente zerlegt und in einem Massenspektrometer gewogen. Im Prinzip verrät das Fragmentmuster, welche Proteinabschnitte räumlich nahe beieinander lagen – ähnlich wie das Finden zusammengeklammerter Seiten eines Buches. In der Praxis sind die resultierenden Daten jedoch extrem komplex. Bestehende Computerwerkzeuge betrachten meist nur die grundlegenden Massendaten und haben Schwierigkeiten mit der enormen Anzahl möglicher Kombinationen, was zu verpassten Verbindungen und falschen Treffern führt.
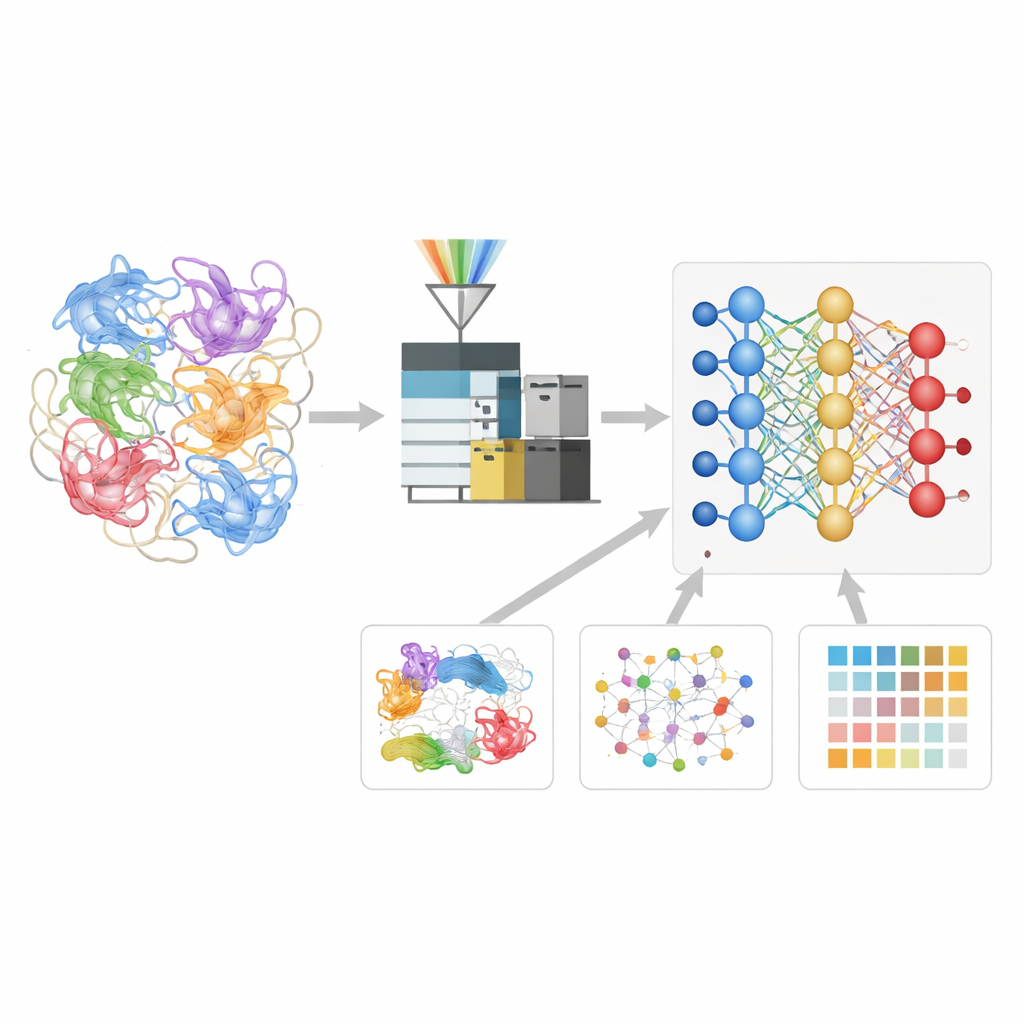
Einem neuronalen Netzwerk die Sprache von Proteinfragmenten beibringen
Die Autoren entwickelten ein tiefes Lernmodell namens Deep4D-XL, um diese Crosslinking-Experimente besser zu interpretieren. Zunächst erstellten sie einen großen Referenzsatz, indem sie Proteine aus menschlichen Zellen vernetzten, sie in Peptide zersetzten und nicht nur ihre Massen, sondern auch ihre Retentionszeiten im Instrument und ihr Verhalten in einer Ionenmobilitätskammer aufzeichneten. Jedes crossvernetzte Paar wurde für das Modell kodiert, das ein Zwillings‑„Siamese“-Design verwendet, um beide Peptidpartner zu lesen, und einen Cross‑Attention-Schritt, um ihre Informationen zu kombinieren. Daraus lernt das Netzwerk, drei Schlüsselmerkmale jedes neuen crossvernetzten Peptids vorherzusagen: wann es im Experiment erscheinen sollte, wie es sich bewegen sollte und wie sein Fragmentierungsmuster aussehen sollte.
Vorhersagen in sauberere Signale verwandeln
XL-MSDigger verpackt diese Vorhersage‑Engine in Analyseworkflows für zwei gängige Datenerfassungsmodi. Im traditionellen, zielgerichteten Modus zeichnet das Gerät selektiv Fragmente von Ionen auf, die es in Echtzeit auswählt. XL-MSDigger nimmt die anfänglichen Treffer etablierter Suchsoftware und bewertet sie erneut anhand des vom Modell prognostizierten Verhaltens für jeden Kandidaten. Ein zweites neuronales Netzwerk vergleicht Vorhersage und Experiment entlang mehrerer Dimensionen und vergibt verbesserte Scores. Dieser Rescoring‑Schritt verdoppelt nahezu die Anzahl der sicher detektierten Verknüpfungen zwischen verschiedenen Proteinen in Hefe‑ und Humanproben, während die Fehlerraten niedrig bleiben, und deckt damit deutlich mehr Protein–Protein‑Interaktionen auf als zuvor.
Sinnvolle Interpretation großer Mengen unvoreingenommener Daten
Eine neuere Art, diese Geräte zu betreiben, die datenunabhängige Akquisition genannt wird, zeichnet Fragmente für nahezu alles in einer Probe auf, was die Abdeckung verbessert, aber überwältigende Datenmengen erzeugt. Bisher gab es keine gute Methode, abzuschätzen, wie viele der resultierenden Crosslinks tatsächlich echt sind. XL-MSDigger nutzt Deep4D-XL, um eine sorgfältig angepasste „Decoy“-Bibliothek falscher Crosslinks zu erstellen, und analysiert dann reale und Decoy‑Einträge gemeinsam. Indem es beobachtet, wie oft Decoys durchrutschen, kann die Software die Falschfund‑Rate abschätzen und ein weiteres neuronales Netzwerk trainieren, um echte von falschen Treffern zu trennen. Dieses Rescoring steigert die Anzahl vertrauenswürdiger crossvernetzter Signale um etwa das Fünffache und erzeugt eine klare Trennung zwischen realen und Decoy‑Mustern.
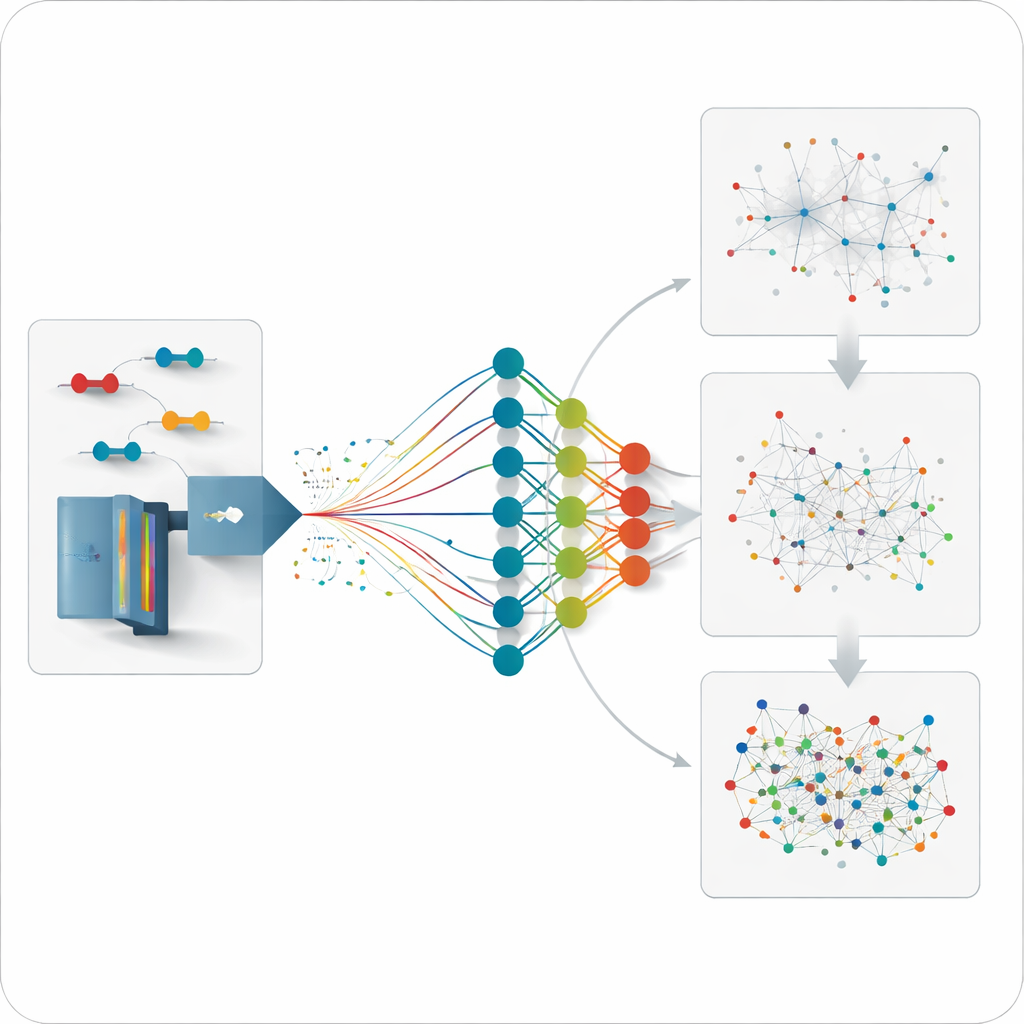
Vorhersagen dessen, was noch nicht gemessen wurde
Da das Modell vorhersagen kann, wie sich jedes plausible crossvernetzte Peptid verhalten sollte, geht das Team noch einen Schritt weiter und analysiert Daten für Verbindungen, die zuvor nie direkt gemessen wurden. Sie erzeugen mittelgroße, vorhergesagte Bibliotheken, die sich auf ausgewählte Proteine oder Interaktionsnetzwerke konzentrieren, und durchsuchen dann die unvoreingenommenen Daten gegen diese Bibliotheken. Diese Strategie deckt zusätzliche Verknüpfungen innerhalb einzelner Proteine und zwischen Partnern wichtiger Chaperon‑Proteine auf, mit Abständen, die gut mit bekannten dreidimensionalen Strukturen übereinstimmen. Sie stellt außerdem Interaktionen wieder her, die von den traditionelleren, eingeschränkteren experimentellen Bibliotheken verpasst wurden, insbesondere bei Verbindungen mit geringer Häufigkeit.
Ein klareres Fenster auf Proteinpartnerschaften öffnen
Für Nicht‑Spezialisten ist die Kernbotschaft, dass XL-MSDigger wie ein hochtrainierter Mustererkenner wirkt, der über einer bereits leistungsfähigen experimentellen Methode liegt. Indem es lernt, wie echte crossvernetzte Signale in mehreren Dimensionen gleichzeitig aussehen sollten, kann es durch gewaltige, unordentliche Datensätze filtern, wahrscheinliche Imitatoren verwerfen und echte, zuvor verborgene Proteinverbindungen wiederfinden. Obwohl Anwendungen für das gesamte Proteom weiterhin erhebliche Rechenressourcen erfordern werden, zeigt diese Arbeit, dass die Kombination von Crosslinking‑Experimenten mit tiefem Lernen unsere Sicht darauf, wie Proteine angeordnet sind und wen sie in der Zelle treffen, deutlich schärfen kann.
Zitation: Chen, M., Hao, Y., Huang, X. et al. XL-MSDigger: a deep learning-based, versatile solution for cross-linking mass spectrometry. Nat Commun 17, 2554 (2026). https://doi.org/10.1038/s41467-026-69489-8
Schlüsselwörter: Proteininteraktionen, Crosslinking-Massenspektrometrie, Tiefes Lernen, Proteomik, datenunabhängige Akquisition