Clear Sky Science · de
Pathophysiologische Bedeutung einer eingeschränkten KAT7‑abhängigen H3K14‑Acetylierung während Zinkmangel
Warum winzige Nährstoffe für unsere Gesundheit wichtig sind
Zink ist ein Spurenelement, das unser Körper nur in winzigen Mengen benötigt, das aber stillschweigend Hunderte von Proteinen unterstützt, die das Zellleben aufrechterhalten. Wenn Zink fehlt – etwa wegen Ernährung, Krankheit oder Alter – wurde das mit Problemen von schlechtem Wachstum über geschwächte Abwehr bis hin zu Fettlebererkrankungen in Verbindung gebracht. Diese Studie stellt eine tiefergehende Frage: Wie merken Zellen eigentlich, dass Zink knapp wird, und wie kann dieser Mangel in nachhaltige Veränderungen der Genaktivität und der Organfunktion umgesetzt werden?
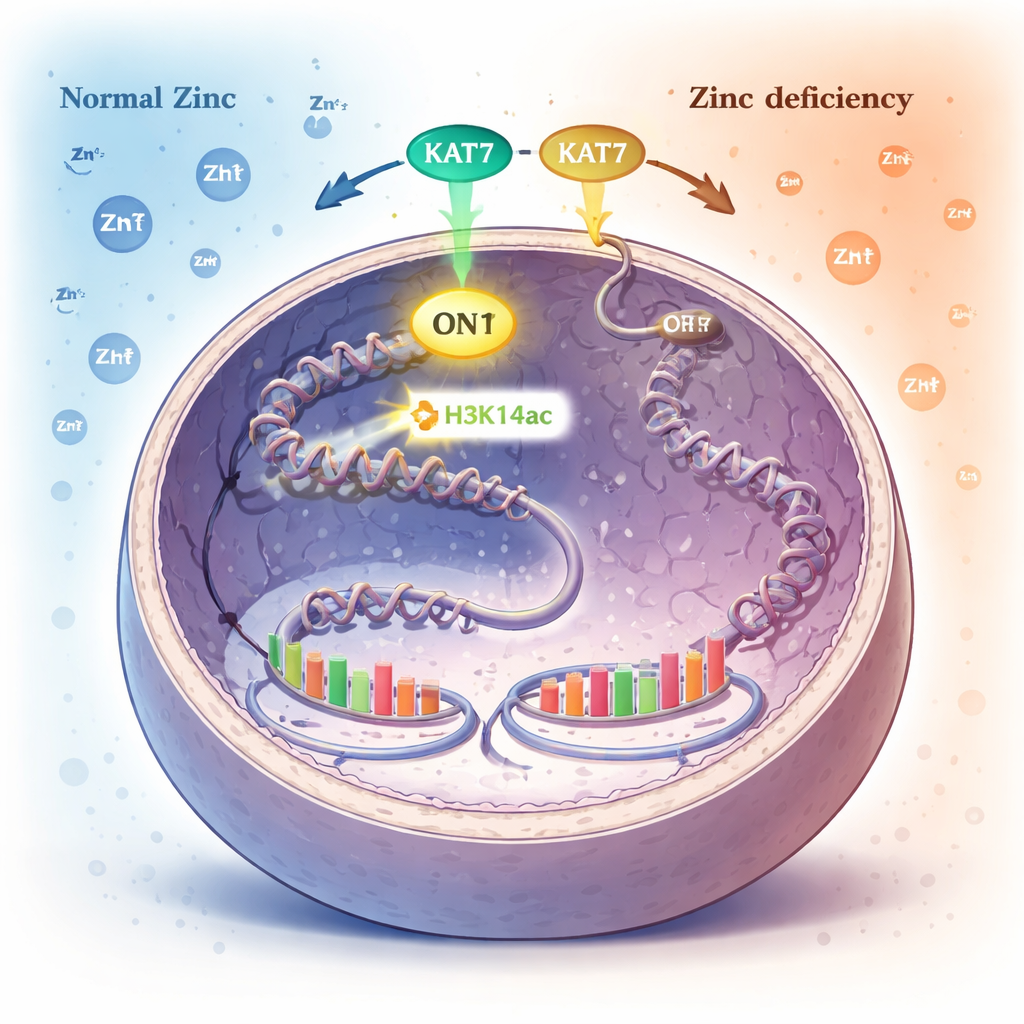
Ein chemisches Zeichen der DNA‑Verpackung als innerer Zinkalarm
Im Zellkern ist DNA um körpereigene „Spulen“ aus Proteinen gewickelt, den Histonen. Zellen steuern, welche Gene aktiv sind, indem sie diesen Histonen kleine chemische Markierungen anheften oder entfernen. Eine solche Markierung, die Acetylierung an einer bestimmten Stelle des Histon H3 (H3K14ac), wird von einem Enzym namens KAT7 angebracht. Die Autorinnen und Autoren fanden heraus, dass bei Zinkknappheit die H3K14ac‑Level drastisch fallen, während viele andere gängige Histonmarken unverändert bleiben. Das deutete darauf hin, dass H3K14ac und das dafür verantwortliche Enzym KAT7 als Schlüsselwächter des Zinkstatus fungieren.
Wie Zink ein zentrales Enzym aktiviert hält
Durch systematisches Deaktivieren verschiedener Enzyme zeigten die Forschenden, dass KAT7 die Hauptquelle von H3K14ac in menschlichen Zellen ist. KAT7 enthält in seinem aktiven Zentrum eine kleine zinkbindende Struktur. Wenn Zellen in Zinkmangel gebracht wurden, nahm KAT7s Fähigkeit, die H3K14ac‑Marke zu setzen, ab, obwohl das Protein selbst weiterhin im Zellkern verblieb und mit seinen Hilfsproteinen assoziiert blieb. Detaillierte Tests mit gereinigten KAT7‑Fragmenten zeigten, dass korrekt gebundenes Zink in diesem Bereich für die Aktivität unerlässlich ist; das Stören der Zinkbindung schaltete das Enzym aus, und das behutsame Ergänzen von Zink stellte die Funktion wieder her. Im Wesentlichen verhält sich KAT7 wie ein zinkabhängiger Schalter, der eine spezifische Histonmarke kontrolliert.
Wie Zinkverlust in Genänderungen übersetzt wird, die Zink wiederherstellen
Was bewirkt der Verlust dieser Histonmarke konkret? Mit genomweiten Kartierungen zeigte das Team, dass H3K14ac besonders an Enhancer‑Regionen angereichert ist – regulatorischen DNA‑Abschnitten, die nahegelegene Gene feinjustieren. Unter zinkarmen Bedingungen wurde H3K14ac von vielen Enhancern entfernt, und je größer der Verlust, desto stärker änderte sich die Aktivität benachbarter Gene. Ein auffälliges Gen war ZIP10, das für ein Protein an der Zelloberfläche kodiert, das Zink importiert. Fiel H3K14ac am Enhancer von ZIP10, stiegen die ZIP10‑Spiegel in der Membran und ermöglichten vermehrten Zinkfluss in die Zelle. Das Blockieren von KAT7 oder das Verhindern des H3K14ac‑Verlusts störte diese Reaktion und verringerte die Zinkaufnahme, sogar nachdem wieder Zink zugeführt wurde. Das zeigt, dass Zellen Zinkmangel in ein epigenetisches Signal umwandeln, das die Zinkimportmaschinerie hochfährt, um das Gleichgewicht wiederherzustellen.
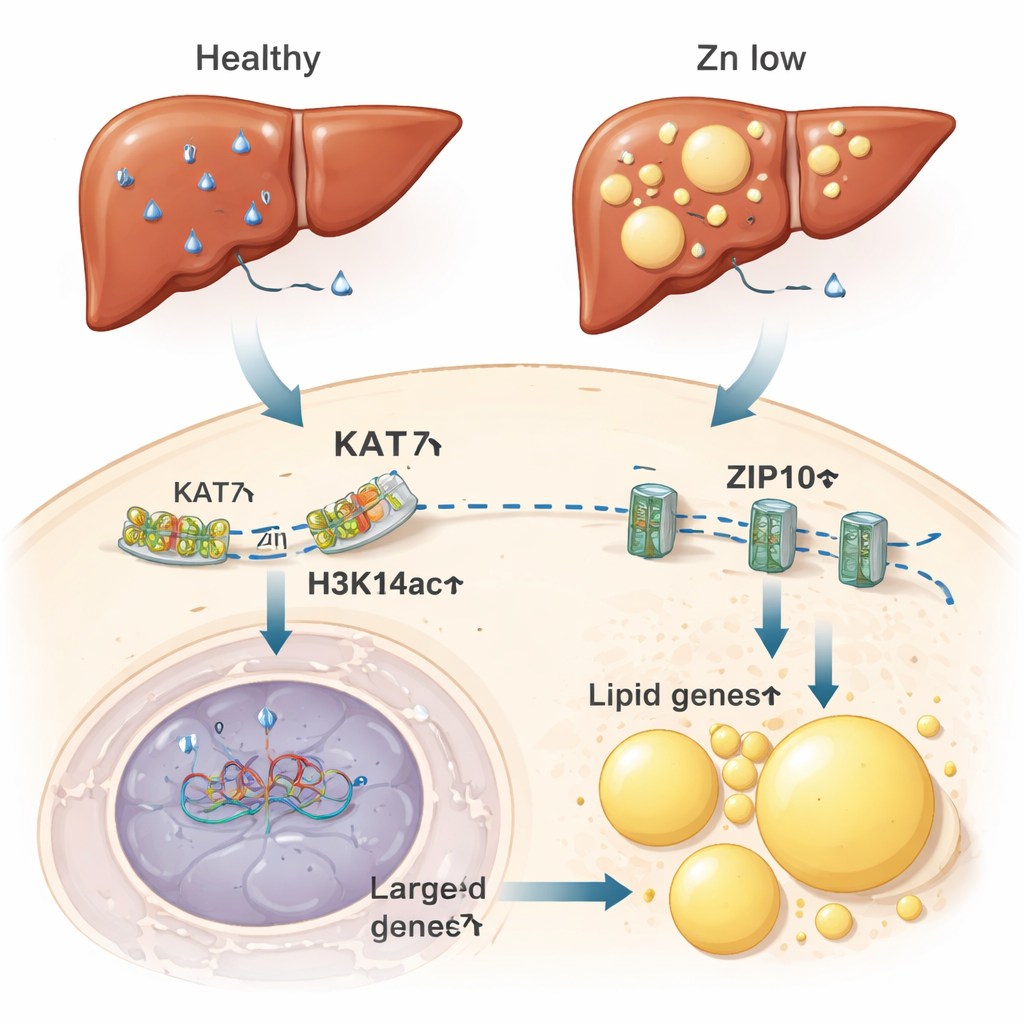
Von zinkentleerten Zellen zur fetten Leber
Die Autorinnen und Autoren fragten anschließend, ob dieser zinksensitive Schalter Folgen im gesamten Organismus hat. Bei Mäusen mit zinkarmer Ernährung zeigten die Lebern – ein zentrales Organ für Zink‑ und Fettstoffwechsel – verringerte Zinkwerte, niedrigere H3K14ac‑Level und verminderte KAT7‑Aktivität. Diese Veränderungen gingen mit einer höheren Expression von Genen einher, die Fettspeicherung und Bildung von Lipidtropfen, den mikroskopischen Fettpaketen in Zellen, antreiben. Die Lebern zinkarmer Mäuse lagerten Fett in einem Ausmaß ein, das mit dem bei einer fettreichen Ernährung vergleichbar war. Bemerkenswerterweise genügte es, die KAT7‑Aktivität pharmakologisch zu senken, ohne die Zinkzufuhr zu ändern, um die Fettansammlung in Leberzellen zu fördern. Umgekehrt verringerte zusätzliche Zinkgabe die durch fettreiche Kost verursachte Fettablagerung.
Was das für das Krankheitsrisiko beim Menschen bedeutet
Um ihre Befunde klinisch einzuordnen, werteten die Forschenden Studien aus, die Zinkwerte im menschlichen Lebergewebe gemessen hatten. In mehreren Berichten wiesen Personen mit Fettleber und verwandten Störungen deutlich weniger Zink in der Leber auf als gesunde Kontrollen. Zusammen mit den Mausdaten legt das nahe, dass chronischer Zinkmangel Fettlebererkrankungen fördern kann, indem er KAT7 stilllegt, die H3K14ac‑Marke ausradiert und dauerhaft Gene hochfährt, die Fettspeicherung begünstigen. Einfach ausgedrückt zeigt die Arbeit einen inneren „Zink‑zu‑Epigenetik“‑Kreislauf: Fällt Zink, verliert ein zinkabhängiges Enzym an Kraft, die DNA‑Verpackung ändert sich und zwar zunächst so, dass Zellen mehr Zink aufnehmen, langfristig aber auch die Leber in Richtung pathologischer Fettansammlung gedrängt werden kann.
Zitation: Fujisawa, T., Takenaka, S., Maekawa, L. et al. Pathophysiological significance of impaired KAT7-dependent histone H3K14 acetylation during zinc deficiency. Nat Commun 17, 1710 (2026). https://doi.org/10.1038/s41467-026-69476-z
Schlüsselwörter: Zinkmangel, Epigenetik, Fettleber, Histonacetylierung, Zinktransporter