Clear Sky Science · de
Eine generische Referenz definiert durch Konsensus-Peaks für die Analyse von Single-Cell-ATAC-seq-Daten
Warum es wichtig ist, die offenen Türen unserer DNA zu kartieren
Jede Zelle in Ihrem Körper trägt im Wesentlichen dieselbe DNA, dennoch verhalten sich Gehirnzellen, Blutzellen und Tumorzellen sehr unterschiedlich. Ein zentraler Grund ist, dass zu einem gegebenen Zeitpunkt nur bestimmte DNA-Abschnitte exponiert und „offen“ für Nutzung sind. Neue Einzelzell-Technologien können diese Offenheit nun genomweit messen, doch fehlte bislang eine gemeinsame Referenzkarte — etwas wie ein Standardatlas — um Ergebnisse zwischen Experimenten und Laboren zu vergleichen. Diese Studie erstellt eine solche Karte, genannt cPeaks, und zeigt, wie sie unsere Sicht auf Zelltypen, Entwicklung und Krebs schärfen kann.
Viele Experimente zu einer gemeinsamen Karte zusammenführen
Die Autoren begannen damit, 624 hochwertige Experimente zu sammeln, die offene Chromatinregionen — die zugänglichen DNA-Teile — in mehr als 40 menschlichen Organen maßen. In jedem Experiment hatten Computerprogramme bereits „Peaks“ markiert, an denen die DNA besonders exponiert war. Anstatt jede Datensatzliste separat zu behandeln, überlagerten die Forscher sorgfältig all diese Peak-Listen entlang des Genoms und verschmolzen sich überlappende Bereiche. Anschließend untersuchten sie, wie oft jede winzige Position innerhalb dieser zusammengefassten Regionen in den Experimenten als offen bezeichnet wurde, und verwandelten jede Region in eine charakteristische Form, die widerspiegelte, wie konsistent sie erschien. Wenn eine zusammengeführte Region tatsächlich mehrere dicht beieinanderliegende offene Stellen enthielt, teilten sie sie in mehrere einfachere Einheiten. Diese Einheiten — insgesamt etwa 1,4 Millionen — wurden zu den beobachteten Konsensus-Peaks, oder cPeaks, einem möglichen Referenzkatalog für menschliche Chromatinzugänglichkeit. 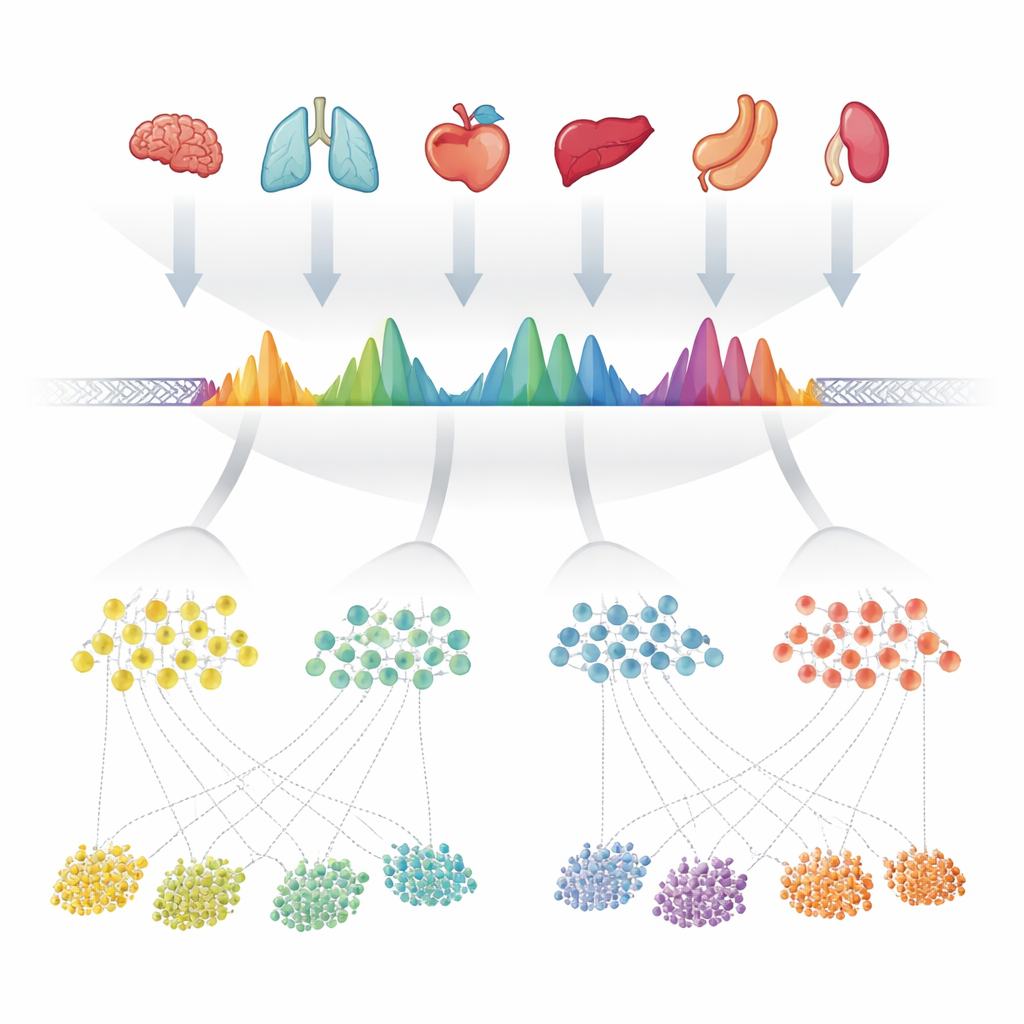
Ein stabiler Fingerabdruck über Gewebe und Technologien hinweg
Damit cPeaks als Referenz nützlich sind, müssen sie echte, reproduzierbare Eigenschaften des Genoms repräsentieren und nicht Eigenheiten bestimmter Proben oder Software. Die Autoren prüften dies, indem sie ihre zusammengefassten Regionen neu erzeugten, wobei sie nur Blutproben, nur feste Gewebe, separate öffentliche Datenbanken und sogar unterschiedliche Labormethoden zur Erfassung offener DNA verwendeten. In jedem Fall ergaben sich an denselben genomischen Positionen bemerkenswert ähnliche Peak-Formen, und die meisten Single-Cell-Datensätze, die sie untersuchten, überlappten mehr als 90 % ihrer eigenen Peaks mit dem cPeak-Katalog. Reads aus vielen Organen sammelten sich präzise um die cPeak-Zentren, was zeigt, dass diese Regionen zuverlässig erfassen, wo Chromatin offen ist. Im Vergleich zu früheren Referenzsätzen, die auf verwandten Technologien basierten, deckten cPeaks mehr der durch ATAC-seq erfassten zugänglichen DNA ab und erfassten nahezu so viel Signal wie frisch in jedem Datensatz definierte Peaks — obwohl sie fest und wiederverwendbar sind.
Ein neuronales Netz beibringen, fehlende Regionen zu finden
Selbst hunderte vorhandener Proben können nicht jeden möglichen Zelltyp abdecken. Um ihre Karte auf bislang nicht beobachtete Regionen auszudehnen, griff das Team auf Deep Learning zurück. Sie trainierten ein eindimensionales Convolutional Neural Network auf DNA-Sequenzen: Beispiele innerhalb beobachteter cPeaks dienten als Positive, während zufällig gewählte Hintergrundregionen als Negative dienten. Das Modell lernte, diese beiden Klassen mit hoher Genauigkeit zu unterscheiden, was darauf hindeutet, dass cPeaks erkennbare Sequenzmuster tragen. Als die Forscher gezielt Peaks entfernen, die jeweils für ein Gewebe spezifisch waren, erholte das Netz diese dennoch allein aus der Sequenz, einschließlich seltener, gewebespezifischer Stellen. Anschließend schoben sie ein kleines Fenster über den Rest des Genoms, bewerteten jedes Segment und fügten etwa 280.000 hochbewertete neue Regionen als prognostizierte cPeaks zum Katalog hinzu, wodurch die Abdeckung insbesondere in ursprünglich unterrepräsentierten Geweben verbessert wurde.
Offene Regionen mit Genen, Zelltypen und seltenen Zellen verknüpfen
Mit einer reicheren Referenz fragten die Autoren, welche Funktionen diese Regionen haben. Viele cPeaks liegen in der Nähe von Genstart- und -endstellen oder überlappen bekannte regulatorische Elemente wie Promotoren, Enhancer und Bindungsstellen für architektonische Proteine wie CTCF. Ein kleiner Teil ist in fast jedem Datensatz zugänglich; diese längeren „Housekeeping“-cPeaks befinden sich tendenziell in Kernpromotorregionen von Genen, die für grundlegende Zellfunktionen notwendig sind. Das Team klassifizierte cPeaks auch danach, wie scharf und konsistent ihre Ränder über Proben hinweg sind, was widerspiegelt, wie präzise die nahegelegene DNA in Nukleosomen verpackt ist. Regionen mit scharf definierten Grenzen sind für bestimmte Familien von Transkriptionsfaktoren angereichert, die dafür bekannt sind, Chromatin umzugestalten und die Entwicklung voranzutreiben. Wenn cPeaks als Merkmalsatz zur Analyse mehrerer Single-Cell-Datensätze verwendet wurden, verbesserten sie die Genauigkeit der Zelltyp-Zuordnung und waren besonders hilfreich beim Identifizieren seltener Zelltypen und subtiler Subtypen, die frühere Peak-Sätze oder einfache genomische Raster oft verwischten.
Entwicklung und Krebs mit einer gemeinsamen Sprache verfolgen
Die Stärke einer Standardreferenz wird deutlich beim Vergleich sehr unterschiedlicher biologischer Kontexte. Mithilfe von cPeaks analysierten die Autoren Single-Cell-Daten der sich entwickelnden menschlichen Retina, große Atlanten fetaler und erwachsener Gewebe sowie mehrere Krebsarten neu. Sie konnten Entwicklungsverläufe rekonstruieren und beobachten, dass der Anteil scharf begrenzter, „gut positionierter“ cPeaks während Übergangsphasen tendenziell ansteigt und dann abfällt, wenn Zellen stabile Identitäten annehmen. Ein ähnliches Muster zeigte sich über Tumorstadien hinweg: Zwischenstadien wiesen einen höheren Anteil dieser strukturierten Regionen auf, was auf intensive regulatorische Umgestaltungen hindeutet. In einem Ovarialtumor halfen cPeaks, zwei unterschiedliche Krebszell-Subklone mit verschiedenen DNA-Kopienzahländerungen zu enthüllen und zeigten, wie die Referenz verborgene Komplexität in Krankheiten offenlegen kann.
Was das für die zukünftige Genomforschung bedeutet
Für Nicht-Spezialisten lassen sich cPeaks als ein standardisiertes Set von Koordinaten vorstellen, das markiert, wo das Genom über viele menschliche Zelltypen hinweg am wahrscheinlichsten physikalisch offen und aktiv ist. Durch Ausrichten neuer Einzelzell-Chromatin-Experimente an dieser gemeinsamen Karte können Forscher Ergebnisse zwischen Studien vergleichen, seltener auftretende oder Übergangszustände von Zellen leichter entdecken und beginnen, großskalige Modelle der Genregulation zu bauen — ähnlich wie standardisierte Genkataloge den Aufstieg von Single-Cell-RNA-Atlanten ermöglichten. Der aktuelle cPeak-Katalog ist ein erster Entwurf, der mit neuen Daten wachsen wird, doch bietet er bereits eine gemeinsame Sprache zur Beschreibung von Chromatinzugänglichkeit und bringt uns einer einheitlichen Sicht darauf näher, wie DNA-Packung Entwicklung, Gesundheit und Krankheit steuert. 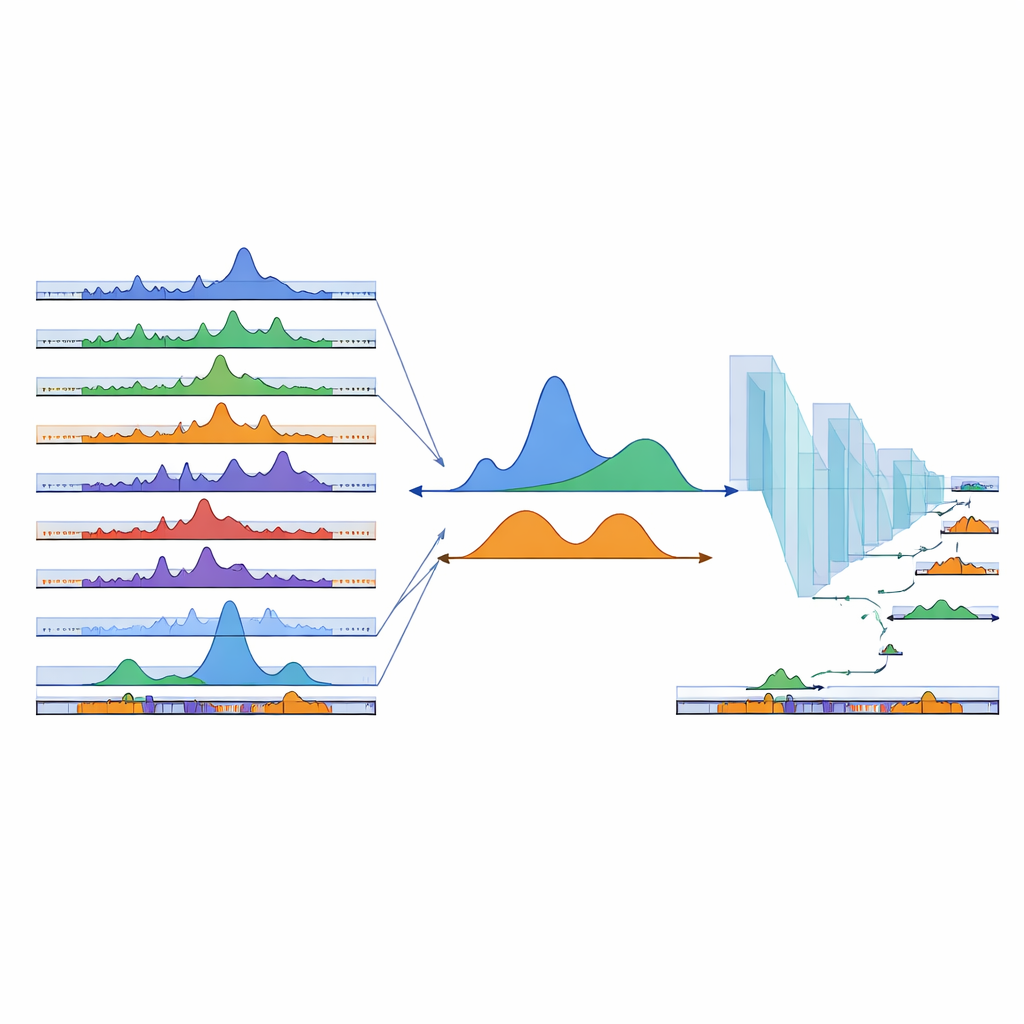
Zitation: Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun 17, 2522 (2026). https://doi.org/10.1038/s41467-026-69461-6
Schlüsselwörter: Chromatinzugänglichkeit, Single-Cell-ATAC-seq, Konsensus-Peaks, Genregulation, Deep-Learning-Genomik