Clear Sky Science · de
CD38 baut MAVS durch Mitophagie ab, um die Sekretion von Typ‑I‑Interferonen in Nasopharynxkarzinom‑Zellen zu hemmen und die CD8+-T‑Zell‑vermittelte Antitumor‑Immunität zu schwächen
Warum das für die Krebsbehandlung wichtig ist
Das Nasopharynxkarzinom ist ein Krebs, der hinter der Nase entsteht und besonders in Ost‑ und Südostasien häufig vorkommt. Immunbasierte Arzneien, die die körpereigenen T‑Zellen aktivieren, haben die Prognose einiger Patientinnen und Patienten verändert, doch die meisten profitieren weiterhin nicht. Diese Studie deckt eine verborgene Bremse innerhalb der Tumorzellen selbst auf: ein Molekül namens CD38, das das interne Alarmsystem stillschweigend abschaltet und dadurch den Angriff krebsabtötender CD8‑T‑Zellen abschwächt. Das Verständnis und die Ausschaltung dieser Bremse könnten bestehende Immuntherapien für viele mehr wirksam machen.
Ein verborgener Schalter auf Tumorzellen
Die Forschenden konzentrierten sich auf CD38, ein Protein, das auf vielen Immunzellen vorkommt, aber auch auf Nasopharynxkarzinom‑Zellen vorhanden ist. Frühere Arbeiten hatten CD38 mit Resistenz gegen gängige Checkpoint‑Medikamente, die PD‑1 und PD‑L1 anvisieren, in Verbindung gebracht. Hier untersuchte das Team, ob CD38 innerhalb der Tumorzellen direkt beeinflusst, wie gut CD8‑T‑Zellen diese Zellen erkennen und zerstören können. Durch Ko‑Kulturen menschlicher Tumorzellen mit oder ohne CD38 zusammen mit aktivierten menschlichen CD8‑T‑Zellen fanden sie heraus, dass das Entfernen von CD38 aus den Krebszellen die T‑Zellen deutlich wirksamer machte: Sie sezernierten höhere Mengen wichtiger Angriffsmediatoren, überlebten besser und töteten mehr Tumorzellen. Wurde CD38 wieder zugegeben, nahm die T‑Zell‑Funktion ab, was auf CD38 als tumorzellintrinsischen Unterdrücker der Immunattacke hinweist.
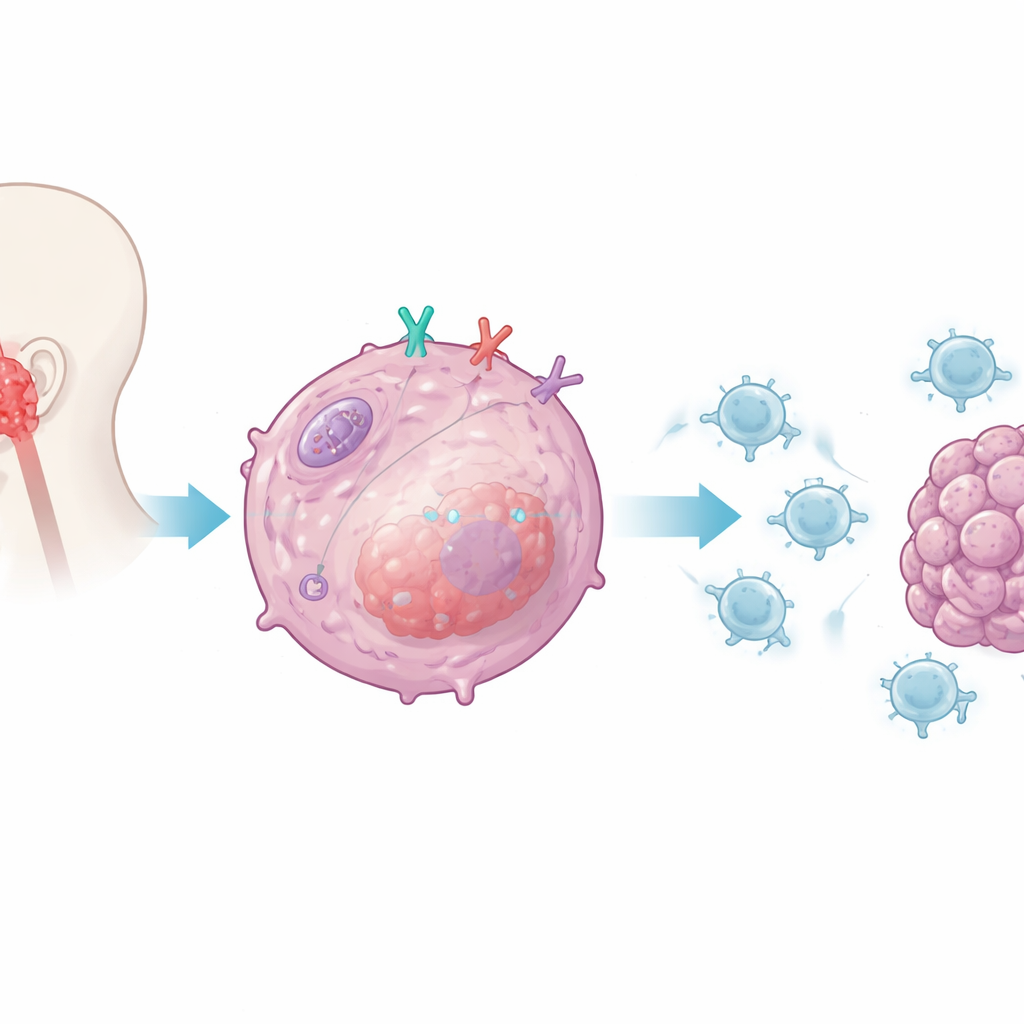
Das interne Alarmsystem der Zelle dämpfen
Das Team untersuchte anschließend, wie CD38 dieses unterdrückende Signal vermittelt. Sie richteten den Blick auf das angeborene Alarmsystem der Tumorzelle, das normalerweise virusähnliche genetische Signale erkennt und Typ‑I‑Interferone auslöst — potente immunstimulierende Botenstoffe. In Tumorzellen ohne CD38 beobachteten die Forschenden einen starken Anstieg von Interferon‑Beta und von Chemokinen, die CD8‑T‑Zellen in Tumoren anlocken. Sie zeigten, dass CD38 selektiv den Weg dämpft, der durch den intrazellulären Sensor RIG‑I und dessen Adapterprotein MAVS gesteuert wird, das auf Mitochondrien sitzt, den Energiestandorten der Zelle. Wenn CD38 vorhanden war, wurde die Aktivierung dieses Weges und seiner nachgelagerten Signalproteine abgeschwächt; wurde CD38 entfernt, stiegen Signalgebung und Interferonproduktion stark an, wodurch die Sichtbarkeit des Tumors für das Immunsystem zunahm.
Wie CD38 einen wichtigen Signalknoten zerstört
Bei genauerer Betrachtung fanden die Wissenschaftlerinnen und Wissenschaftler, dass CD38 physisch mit MAVS auf Mitochondrien assoziiert und MAVS’ Partnerschaft mit RIG‑I stört, wodurch die Signalübertragung geschwächt wird. Auffälliger noch: Höhere CD38‑Spiegel führten zu einer Verringerung des MAVS‑Proteins, ohne dessen genetische Vorlage zu verändern, was auf aktiven Abbau hindeutet. Tests mit verschiedenen Inhibitoren zeigten, dass dieser Verlust von der Recycling‑Maschinerie der Zelle, der Autophagie, abhängt — und speziell von einer Form, die Mitochondrien gezielt entfernt. CD38 erhöhte Marker für mitochondriales „Selbstfressen“, reduzierte mehrere mitochondriale Proteine und förderte die Einlagerung von MAVS in Autophagosom‑Strukturen, die später abgebaut werden. Das Blockieren der mitochondrialen Autophagie bewahrte MAVS und stellte die Interferon‑Signalgebung wieder her, was darauf hindeutet, dass CD38 den Alarm abschaltet, indem es MAVS in den zellulären Abfallweg treibt.
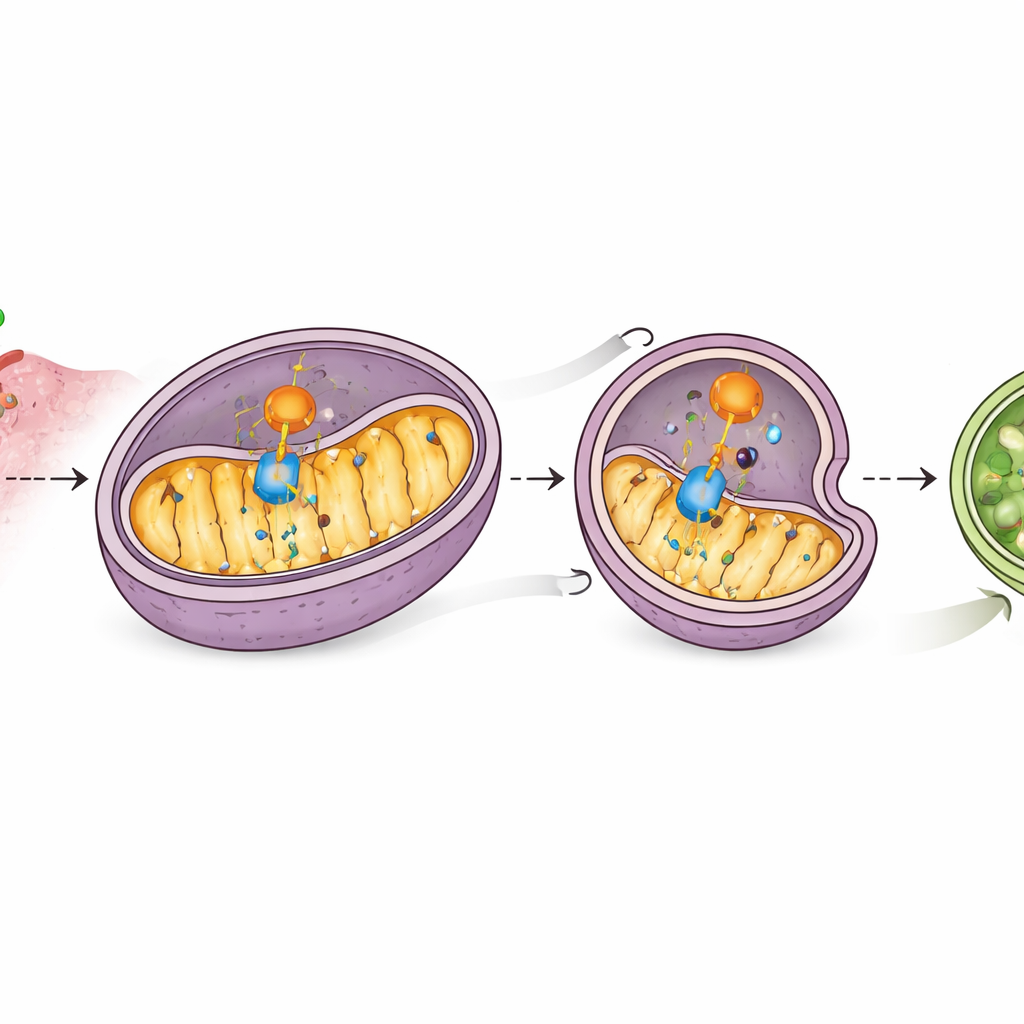
Ein Helfer, der Mitochondrien zur Selbstzerstörung führt
Die Studie identifizierte einen weiteren Akteur, PHB2, ein Protein innerhalb der Mitochondrien, das als Rezeptor für zielgerichtete mitochondriale Entfernung fungiert. Massenspektrometrie und Bindungsexperimente zeigten, dass CD38 mit PHB2 interagiert und PHB2s Präsenz in Mitochondrien erhöht, wo PHB2 wiederum die Kernmaschinerie der Autophagie rekrutiert. PHB2 bindet auch MAVS, und CD38 verstärkt diesen Kontakt. Wurde PHB2 reduziert, konnte CD38 die mitochondriale Autophagie nicht mehr effektiv auslösen, die MAVS‑Spiegel erholten sich und interferonbezogene Gene wurden reaktiviert. Das offenbart eine Abfolge: CD38 bindet PHB2, PHB2 bindet MAVS, und gemeinsam leiten sie MAVS in Mitochondrien, die dem Abbau zugeführt werden, und schweigen so den Interferon‑Alarm.
Belege aus Tiermodellen
Um die Auswirkungen in lebenden Organismen zu testen, nutzten die Forschenden Maus‑Tumoren, bei denen CD38 ausgeschaltet war. In immunkompetenten Mäusen wuchsen diese Tumoren langsamer, enthielten mehr CD8‑T‑Zellen und hatten einen höheren Anteil an Zellen, die Interferon‑Gamma produzierten — ein Kennzeichen aktiver Antitumor‑Antworten. Die Blockade des Rezeptors für Typ‑I‑Interferone nahm diesen Vorteil weg und bestätigte, dass Interferon‑Signalgebung für die verbesserte Immunität wesentlich ist. In humanisierten Mäusen mit Nasopharynx‑Tumoren verlangsamte die Reduktion von CD38 ebenfalls das Tumorwachstum und erhöhte die CD8‑T‑Zell‑Infiltration, doch dieser Nutzen verschwand, wenn MAVS in den Tumorzellen gleichzeitig reduziert wurde. Diese in vivo Beobachtungen untermauern die Idee, dass die CD38–PHB2–MAVS‑Achse innerhalb der Tumorzellen die Stärke der T‑Zell‑Antwort des Körpers bestimmt.
Was das für künftige Therapien bedeutet
Insgesamt zeigt die Arbeit, dass CD38 in Nasopharynxkarzinom‑Zellen als interner Saboteur der Antitumor‑Immunität wirkt. Durch das Auslösen einer selektiven Form der mitochondrialen Rekyclingprozesse entleert CD38 MAVS, schwächt die Produktion von Typ‑I‑Interferonen, reduziert die Antigenpräsentation und dämpft letztlich den Angriff von CD8‑T‑Zellen. Aktuelle CD38‑blockierende Verbindungen richten sich hauptsächlich gegen seine Enzymaktivität und entfernen das Protein nicht oder stellen MAVS nicht wieder her. Die Autorinnen und Autoren argumentieren, dass neue Strategien, die darauf abzielen, CD38‑Spiegel zu reduzieren oder seine Partnerschaft mit PHB2 oder MAVS zu stören, den Interferon‑Alarm innerhalb von Tumoren reaktivieren könnten. In Kombination mit bestehenden Checkpoint‑Inhibitoren könnten solche Ansätze mehr Nasopharynx‑ und möglicherweise auch andere Tumoren von immun‑kalt in immun‑reaktiv verwandeln.
Zitation: Liang, L., Li, W., Liu, S. et al. CD38 degrades MAVS through mitophagy to inhibit type I interferon secretion in nasopharyngeal carcinoma cells and impairs CD8+T cell-mediated anti-tumor immunity. Nat Commun 17, 2544 (2026). https://doi.org/10.1038/s41467-026-69339-7
Schlüsselwörter: Nasopharynxkarzinom, Tumorimmuntherapie, Typ‑I‑Interferon, CD8‑T‑Zellen, Mitophagie