Clear Sky Science · de
Loss-of-function-Varianten im CAPN1-Aktivator CD99L2 verursachen X‑chromosomal vererbte spastische Ataxie
Warum das für Familien mit unerklärten Bewegungsstörungen wichtig ist
Viele Menschen leben jahrelang mit unerklärlichen Gehproblemen, Muskelsteifigkeit oder Schwierigkeiten mit Gleichgewicht und Sprache, ohne jemals die tatsächliche Ursache zu erfahren. Diese Studie zeigt, wie moderne DNA‑Tests einigen dieser Familien endlich eine Antwort liefern können. Die Forschenden verglichen nicht nur verschiedene genetische Tests für seltene Bewegungsstörungen, sondern entdeckten auch eine zuvor unbekannte Ursache einer als X‑chromosomal vererbte spastische Ataxie, was auf biologische Signalwege hinweist, die auch bei häufigeren Erkrankungen des Gehirns eine Rolle spielen könnten.
Die genetische Nadel im Heuhaufen seltener Krankheiten finden
Seltene Bewegungsstörungen wie Ataxie (ungewollt unsichere Bewegungen) und spastische Paraplegie (steife, schwache Beine) werden oft als genetisch vermutet, doch bei den meisten Patientinnen und Patienten liefern Standardtests keine Erklärung. Das Team begleitete über sechs Jahre 2.811 Personen in Deutschland und Europa, die wegen des Verdachts auf eine seltene Bewegungsstörung überwiesen wurden. Zunächst wurden klassische gezielte Tests durchgeführt, die nach bekannten Repeat‑Expansionen in wenigen Genen suchen; diese lieferten in etwa 11 % der Fälle eine Antwort. Anschließend nutzten die Forschenden Exomsequenzierung, die nur die proteincodierenden Teile des Genoms liest, und fanden eindeutige genetische Erklärungen in etwa 19 % der Fälle, insbesondere bei Personen mit Spastizität.
Über die Standardtests hinaus: Ganzgenomsequenzierung
Um weiter vorzudringen, verwendeten die Wissenschaftlerinnen und Wissenschaftler Ganzgenomsequenzierung, die nahezu die gesamte DNA einer Person erfasst, einschließlich Regionen, die Standardtests und Exome übersehen können. Unter 486 Personen, die diesen umfassenderen Test erhielten, stieg die Diagnoserate um etwa 7,5 Prozentpunkte, vor allem weil die Genomsequenzierung komplexe Veränderungen wie strukturelle Umlagerungen und Repeat‑Expansionen besser erkennt. Die Studie zeigte außerdem, dass sorgfältig dokumentierte klinische Informationen – insbesondere genaue Symptombeschreibungen, jüngeres Alter zum Zeitpunkt der Untersuchung und die Kombination von Spastizität mit weiteren Bewegungsstörungen – vorhersagten, wer mit höherer Wahrscheinlichkeit eine klare genetische Diagnose erhielt.
Aufdeckung einer neuen X‑chromosomalen Ursache spastischer Ataxie
Selbst nach diesen umfangreichen Tests blieben viele Patientinnen und Patienten ohne Diagnose. Die Forschenden bündelten genetische Daten von mehr als 13.000 Personen und nutzten einen sogenannten Gen‑Burden‑Ansatz, um zu prüfen, welche Gene bei Betroffenen häufiger verdächtige Varianten tragen als bei unbetroffenen Kontrollen. Diese Analyse wies nicht nur auf bekannte Krankheitsgene hin, sondern hob auch stark ein bislang vernachlässigtes Gen auf dem X‑Chromosom namens CD99L2 hervor. Durch die Zusammenführung von Ergebnissen aus mehreren Familien in Europa identifizierten sie 25 betroffene Männer aus 20 Familien, die schädigende Varianten in diesem Gen trugen. Diese Männer entwickelten typischerweise im mittleren bis höheren Erwachsenenalter Gehprobleme, Beinsteifigkeit, verwaschene Sprache und manchmal Gleichgewichtsstörungen, während weibliche Trägerinnen größtenteils unbeeinträchtigt blieben – ein Muster, das zu einer X‑chromosomal vererbten Erkrankung passt. Die Varianten zerstörten meist das normale Protein oder entfernten entscheidende Teile davon, was stark darauf hindeutet, dass ein Funktionsverlust die Erkrankung verursacht.
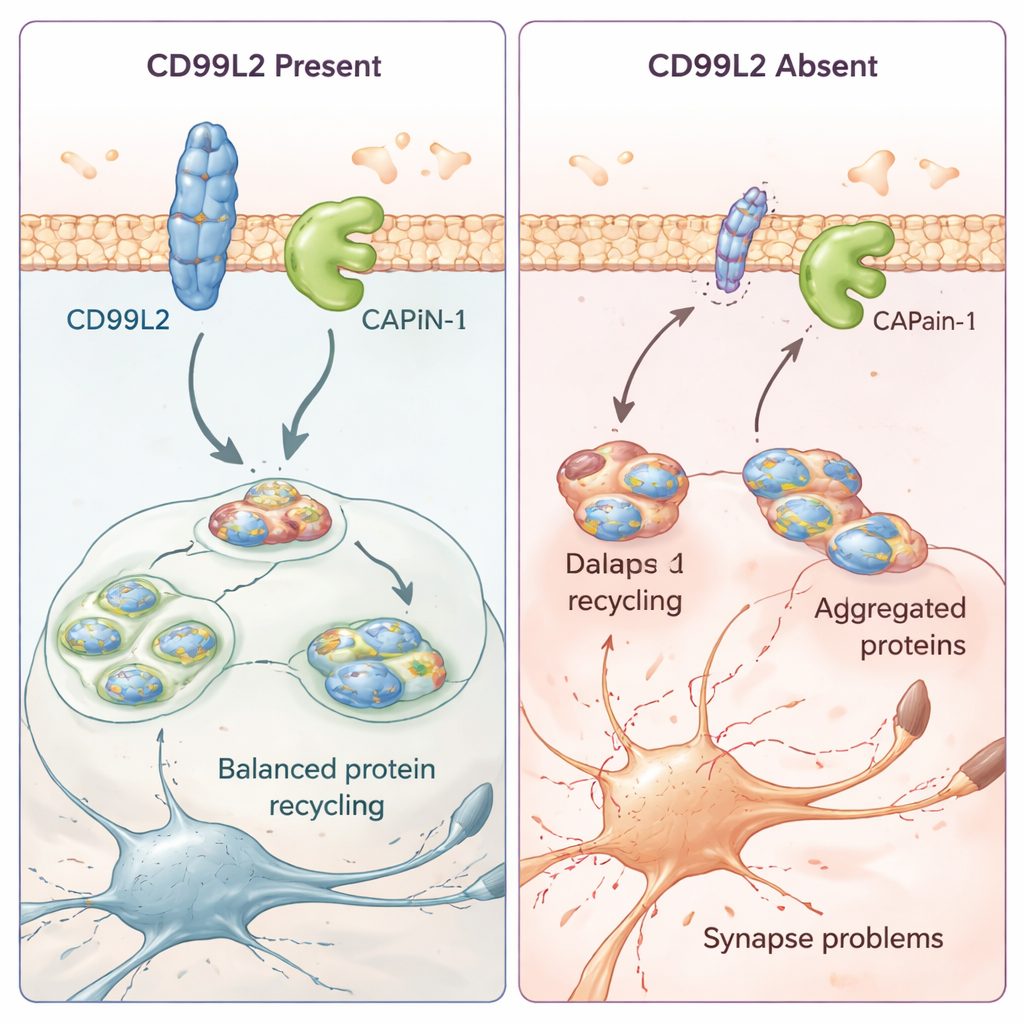
Wie ein kleines Membranprotein Gehirnzellen schützt
Um zu verstehen, welche Rolle CD99L2 in Zellen spielt, arbeiteten die Forschenden mit Zellmodellen und patienteneigenen Hautzellen. Sie fanden heraus, dass das CD99L2‑Protein in der Zellmembran sitzt und üblicherweise mit kleinen „Ubiquitin“‑Markern versehen ist, die steuern, wie lange es bestehen bleibt, bevor es abgebaut wird. CD99L2 bindet physisch an Calpain‑1 (CAPN1), ein calciumaktiviertes Enzym, das andere Proteine beschneidet und dazu beiträgt, Synapsen – die Kontaktstellen zwischen Nervenzellen – gesund zu erhalten. Wenn CD99L2 vorhanden und intakt ist, hilft es, Calpain‑1 kontrolliert ein‑ und auszuschalten, und wird dann selbst zugeschnitten und recycelt. Fehlt CD99L2 oder ist es strukturell verändert, ist die Aktivierung von Calpain‑1 gestört. In Patienten‑Zellen geht das einher mit veränderten Aktivitätsmustern vieler Gene, die mit Synapsen und der Kommunikation von Nervenzellen zu tun haben, was darauf hindeutet, dass subtile, aber weitreichende Veränderungen in der Gehirnverdrahtung den schrittweisen Funktionsverlust und die Bewegungsprobleme erklären könnten.
Was das für Patientinnen und Patienten heute und künftig bedeutet
Für Familien mit unerklärter spastischer Ataxie oder spastischer Paraplegie bringt diese Arbeit zwei Fortschritte. Erstens zeigt sie, dass der frühzeitige Einsatz von Ganzgenomsequenzierung zusammen mit einer sorgfältigen klinischen Erfassung die Chancen auf eine gesicherte genetische Diagnose deutlich erhöhen kann. Zweitens ergänzt sie CD99L2 die Liste der Gene, die die Calpain‑Aktivität steuern – ein Signalweg, der bereits mit anderen seltenen Ataxien und mit häufigen Erkrankungen wie Alzheimer und Parkinson in Verbindung gebracht wurde. Anschaulich: Die Studie beschreibt einen neuen „Ein‑/Aus‑Schalter“, der die Wartung von Gehirnzellen im Gleichgewicht hält; wenn dieser Schalter defekt ist, gehen Nervenzellen langsam zugrunde, was zu Steifheit und Koordinationsstörungen führt. Das Verständnis dieses Schalters könnte langfristig den Weg zu Therapien öffnen, die die Calpain‑Aktivität feinjustieren und Gehirnzellen bei verschiedenen neurologischen Erkrankungen schützen.
Zitation: Menden, B., Incebacak Eltemur, R.D., Demidov, G. et al. Loss-of-function variants in the CAPN1 activator CD99L2 cause X-linked spastic ataxia. Nat Commun 17, 1698 (2026). https://doi.org/10.1038/s41467-026-69337-9
Schlüsselwörter: spastische Ataxie, seltene Bewegungsstörungen, Genomsequenzierung, CD99L2, Calpain‑1