Clear Sky Science · de
Gezielte Hemmung von NHEJ aktiviert STING‑Signalgebung durch MYC‑Abbau und stärkt die antitumorale Immunität bei SCLC
Warum diese Forschung wichtig ist
Das kleinzellige Lungenkarzinom gehört zu den tödlichsten Krebsarten; die meisten Patientinnen und Patienten überleben nach der Diagnose weniger als ein Jahr. Auffällig ist, dass diese Tumoren viele DNA‑Mutationen aufweisen, die sie theoretisch zu guten Zielen für das Immunsystem machen sollten, in der Praxis aber schlecht auf moderne Immuntherapien ansprechen. Diese Studie entlarvt eine bisher verborgene molekulare Bremse, die das Immunsystem daran hindert, diese Tumoren zu erkennen, und zeigt, wie das Abschalten eines zentralen DNA‑Reparaturproteins diese Tumoren von immunologisch "kalt" zu "heiß" umschalten kann, sodass bestehende Therapien deutlich wirksamer werden.
Ein versteckter Reparaturschalter in Lungentumoren
Die Forschenden begannen mit der Auswertung genetischer Daten aus mehr als 179.000 menschlichen Tumoren aus 24 Krebsarten. Sie konzentrierten sich auf einen DNA‑Reparaturweg namens nicht‑homologe End‑Verknüpfung (NHEJ), der gefährliche Brüche in DNA‑Strängen schließt. Ein zentraler Regulator dieses Wegs, ein Protein namens DNAPKcs (kodiert durch das PRKDC‑Gen), erwies sich bei kleinzelligem Lungenkrebs als ungewöhnlich hoch. Unter tausenden Lungentumorproben zeigten die kleinzelligen Fälle die stärkste Aktivität dieses Reparaturschalters. Patientinnen und Patienten, deren Tumoren hohe PRKDC‑Spiegel aufwiesen, lebten kürzer und profitierten seltener von Standard‑Chemotherapien und Checkpoint‑Inhibitoren, was nahelegt, dass DNAPKcs den Tumoren hilft, sowohl DNA‑Schäden als auch Immunangriffen zu widerstehen.
Von DNA‑Schaden zu einem inneren Alarm
Um zu untersuchen, was passiert, wenn dieser Reparaturschalter abgeschaltet wird, setzten die Forscherinnen und Forscher sowohl Medikamente als auch genetische Stilllegungswerkzeuge ein, um DNAPKcs in Zelllinien des kleinzelligen Lungenkrebses und in Maus‑Tumormodellen zu blockieren. In vielen Modellen, besonders in solchen, die menschlichen Subtypen mit hoher MYC‑Onkogenaktivität ähneln, führten DNAPKcs‑Inhibitoren zu einer ausgeprägten Verringerung des Tumorzellwachstums und schrumpften sogar patientenabgeleitete Tumoren in Mäusen. Auf zellulärer Ebene führte die Blockade von DNAPKcs zur Anhäufung gebrochener DNA, sichtbar als Punkte eines Schadensmarkers im Zellkern und als winzige, DNA‑gefüllte Körperchen (Mikronuklei). Diese DNA‑Fragmente gelangten ins Cytoplasma, wo sie als Gefahrensignale wahrgenommen werden konnten.
Aktivierung des zellulären "Virus"‑Alarmsystems
Lose DNA an der falschen Stelle ist normalerweise ein Hinweis auf eine Virusinfektion. Zellen erkennen sie mithilfe eines Sensors namens cGAS, der einen nachgeschalteten Alarmweg namens STING auslöst. Die Autorinnen und Autoren zeigten, dass nach DNAPKcs‑Hemmung cGAS an Mikronuklei aggregierte, STING aktiviert wurde und eine Kaskade immunstimulierender Moleküle in Gang gesetzt wurde. Die Zellen produzierten mehr Typ‑I‑ und Typ‑II‑Interferone sowie Chemokine, die Immunzellen anziehen. Auch die Oberflächenpräsentation wichtiger "Marke"‑Proteine (MHC‑Klasse‑I‑Moleküle), die Immunzellen helfen, Tumorantigene zu erkennen, nahm zu. Wurde der STING‑Weg chemisch blockiert oder genetisch ausgeschaltet, verschwanden diese Veränderungen größtenteils, und die antitumoralen Effekte der DNAPKcs‑Hemmung waren deutlich abgeschwächt – ein Hinweis darauf, dass dieses interne Alarmsystem für die Reaktion essenziell ist.
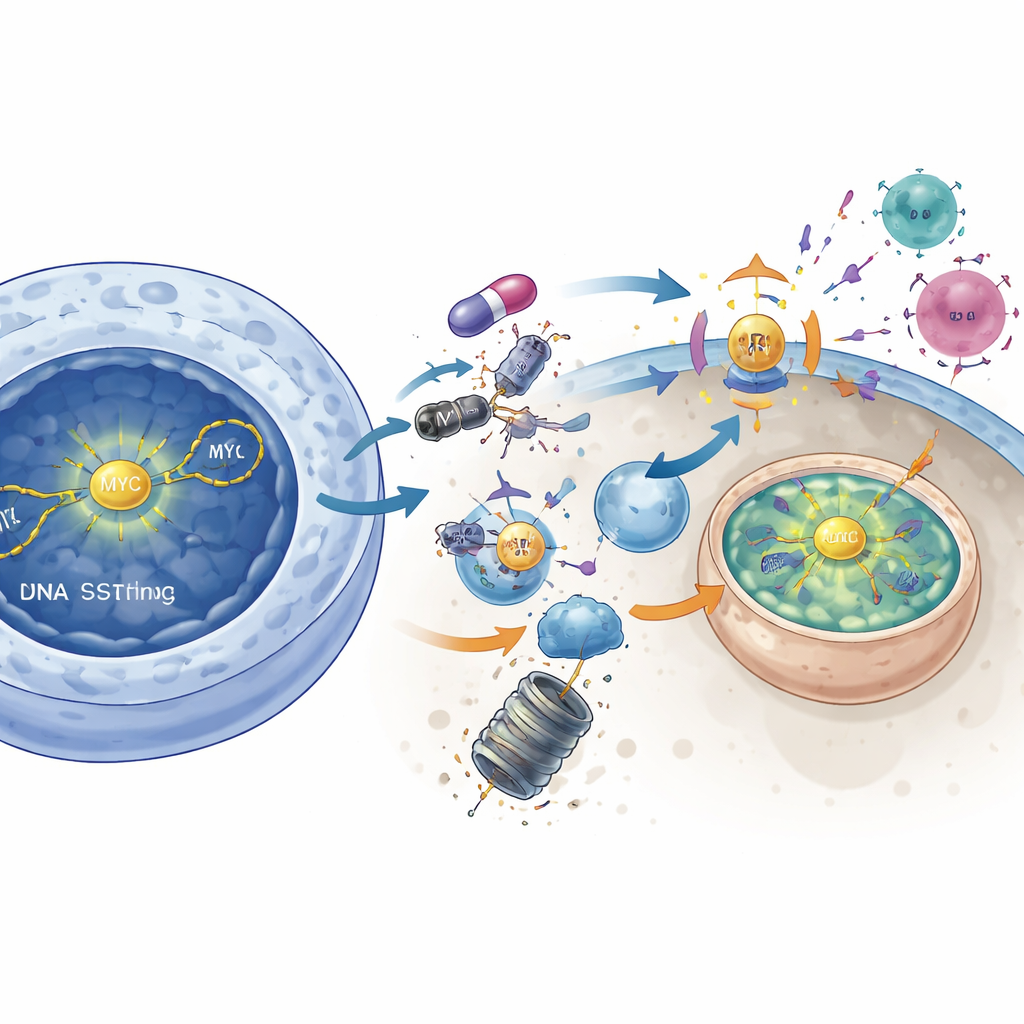
MYC entwaffnen, um den Tumor sichtbar zu machen
Die Studie verbindet DNAPKcs außerdem mit dem starken Wachstumsförderer MYC, einem lange als "unverdaubaren" Angriffspunkt geltenden Protein. In Tumoren mit hoher MYC‑Aktivität reduzierte die DNAPKcs‑Blockade aktive AKT‑Signalgebung und löste eine molekulare Bremse an einem anderen Enzym, GSK3β. Nach dessen Aktivierung markierte GSK3β MYC für den Abbau, wodurch die MYC‑Proteinspiegel absanken. Ein direkter genetischer Abbau von MYC ahmte viele der immunaktivierenden Effekte der DNAPKcs‑Blockade nach: STING‑Signalgebung stieg, Interferongene wurden eingeschaltet und MHC‑Klasse‑I nahm zu. Umgekehrt tilgte eine erzwungene Überexpression von MYC größtenteils den immunstärkenden Effekt des DNAPKcs‑Inhibitors. Das spricht dafür, dass DNAPKcs normalerweise zur Stabilisierung von MYC beiträgt und dass die Induktion von MYC‑Abbau ein entscheidender Schritt ist, um die antitumorale Immunität zu reaktivieren.
Von "kalten" zu "heißen" Tumoren in lebenden Modellen
In immunintakten Mausmodellen, die dem menschlichen kleinzelligen Lungenkrebs nahekommen, verlangsamte oder verkleinerte eine Behandlung mit einem DNAPKcs‑Inhibitor allein die Tumoren deutlich. Wichtig ist, dass die Kombination des Inhibitors mit einem bestehenden Anti‑PD‑L1‑Checkpoint‑Medikament zuvor resistente Tumoren verwandelte, was zu dramatischen Tumorregressionen und in einigen Fällen zum vollständigen Verschwinden führte. Detaillierte immunologische Profilierungen zeigten, dass die DNAPKcs‑Hemmung krebsabtötende CD8‑T‑Zellen vermehrte, proinflammatorische M1‑Makrophagen stärkte, unterdrückende T‑Zellen reduzierte und die MHC‑Klasse‑I‑Expression in Tumoren erhöhte. Das Entfernen von CD8‑T‑Zellen oder das Deaktivieren von STING hob diese Vorteile wieder auf, was bestätigt, dass die Therapie wirkt, indem sie den Tumor zu einem Leuchtfeuer für Immunangriffe macht und nicht nur durch direkte Krebszelltötung.
Was das für Patientinnen und Patienten bedeutet
In der Summe identifizieren diese Ergebnisse DNAPKcs als zentralen Koordinator sowohl der DNA‑Reparatur als auch der Immun‑Evasion beim kleinzelligen Lungenkrebs. Durch die Blockade von DNAPKcs sammeln sich DNA‑Schäden an, MYC wird destabilisiert, der cGAS–STING‑Alarm wird ausgelöst und Interferon‑ sowie antigenpräsentierende Wege werden aktiviert. Diese Ereigniskette verwandelt immunologisch stille Tumoren in solche, die in präklinischen Modellen stark auf Checkpoint‑Blockade und Chemotherapie ansprechen. Klinische Studien sind zwar noch erforderlich, doch deuten die Ergebnisse darauf hin, dass vorhandene DNAPKcs‑Inhibitoren mit Immuntherapien kombiniert werden könnten, um Patientinnen und Patienten mit diesem aggressiven Krebs bessere Chancen auf eine dauerhafte Kontrolle zu geben.
Zitation: Chakraborty, S., Elliott, A., Sen, U. et al. Targeting NHEJ activates STING signaling through MYC degradation to boost antitumor immunity in SCLC. Nat Commun 17, 2597 (2026). https://doi.org/10.1038/s41467-026-69262-x
Schlüsselwörter: kleinzelliges Lungenkarzinom, Hemmung der DNA‑Reparatur, STING‑Signalweg, MYC‑Abbau, Tumorimmuntherapie