Clear Sky Science · de
Können ferric‑oxyl angeregte Zustände verlängerte Eisen‑Sauerstoff‑Bindungen in katalytischen Intermediaten von Häm‑Peroxidasen erklären?
Warum Eisen‑Sauerstoff‑Bindungen in Enzymen wichtig sind
In unseren Zellen nutzen spezielle Proteine, sogenannte Enzyme, Sauerstoff, um kraftvolle chemische Reaktionen sicher durchzuführen. Unter ihnen verlassen sich Häm‑Peroxidasen auf ein Eisen–Sauerstoff‑Paar in ihrem Kern, um Wasserstoffperoxid abzubauen, ein reaktives und potenziell schädliches Molekül. Jahrzehntelang waren sich Wissenschaftler über die genaue Natur dieser Eisen–Sauerstoff‑Bindung uneinig: Handelt es sich eher um eine enge Doppelbindung oder um eine lockerere Einfachbindung — und was bedeutet das für die Funktionsweise dieser Enzyme? Diese Studie geht dem Rätsel mit ultrakurzen Röntgenmethoden und fortgeschrittenen Rechnungen nach und zeigt, dass die Antwort in kurzlebigen angeregten Zuständen der Eisen–Sauerstoff‑Einheit selbst liegt.
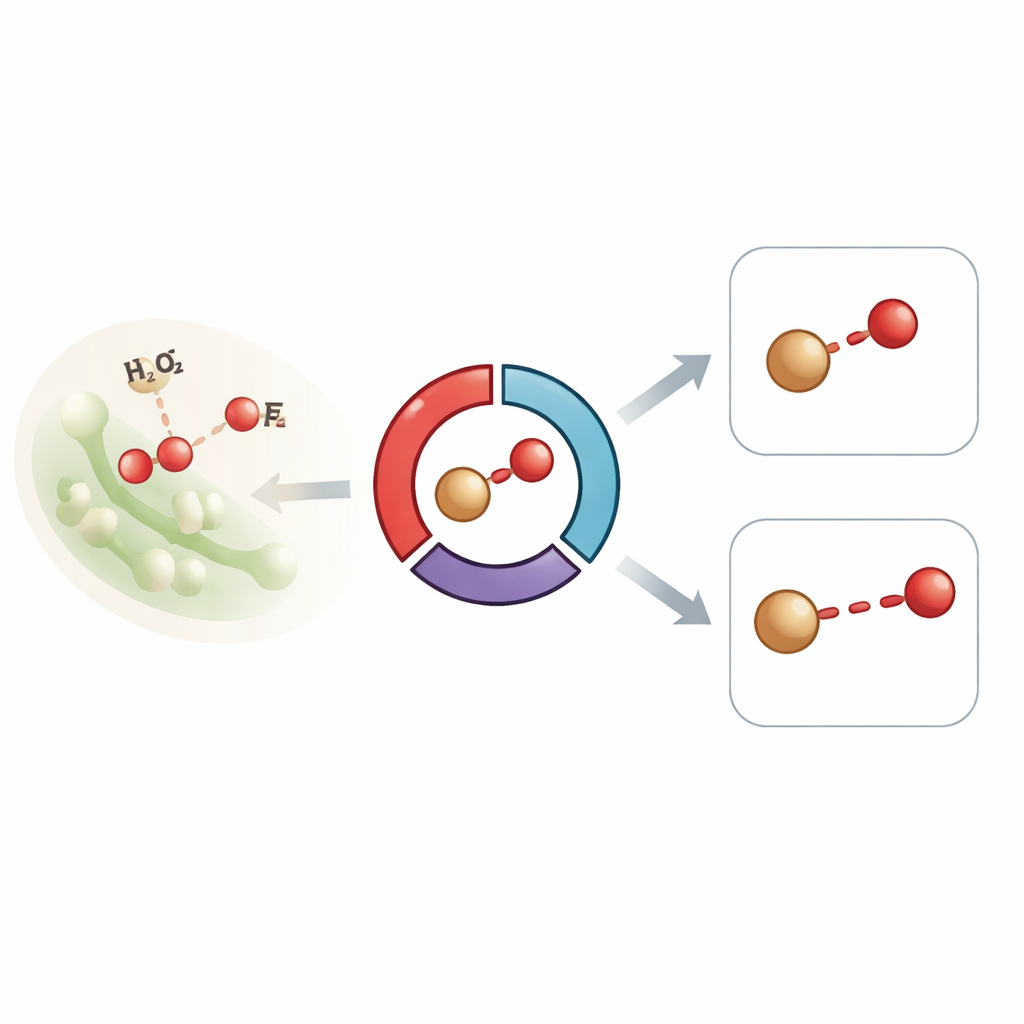
Ein Enzym in Echtzeit verfolgen
Die Forscher konzentrierten sich auf eine bakterielle Dye‑Decolourising‑Peroxidase, ein Häm‑Enzym, das gewöhnlich durch zwei wichtige energiegeladene Formen zyklisiert, bekannt als Compound I und Compound II. Diese Formen weisen beide ein an Sauerstoff gebundenes Eisenatom auf und sind zentral dafür, wie das Enzym Wasserstoffperoxid verarbeitet und andere Moleküle oxidiert. Frühere Experimente an ähnlichen Enzymen zeigten rätselhafterweise lange Eisen–Sauerstoff‑Abstände, die einige Wissenschaftler als Hinweis darauf deuteten, dass die vermeintlich hochreaktive Eisen–Sauerstoff‑Einheit entweder durch Röntgenstrahlung verändert worden war oder ein zusätzliches Proton aufgenommen hatte, wodurch sich ihr Charakter änderte. Um solche Artefakte zu vermeiden, nutzte das Team zeitaufgelöste serielle Femtosekunden‑Röntgenkristallographie an einem X‑Ray‑Free‑Electron‑Laser und erfasste Beugungs‑ und Röntgen‑Emissionssignale von Tausenden winziger Proteinkristalle bei Raumtemperatur, alles innerhalb von Zehnern von Femtosekunden — schneller als Schäden auftreten können.
Die Chemie innerhalb der Kristalle beobachten
In ihrem Aufbau wurden Mikrokristalle einer leicht modifizierten Version des Enzyms direkt auf einem sich bewegenden Band mit Wasserstoffperoxid gemischt und dann nach Verzögerungen von einer halben Sekunde bis zu mehreren Minuten untersucht. Frühe Zeitpunkte begünstigen die Bildung von Compound I, spätere werden von Compound II dominiert. Strukturdaten zeigten, dass sich in beiden Intermediaten das Eisenatom neben einem einzigen Sauerstoffatom in der Hämtasche befindet und dass sich schützende Schleifenbereiche des Proteins verschieben, um dieses stark oxidierende Zentrum abzuschirmen. Wichtig ist, dass präzise Messungen zeigten, dass die Eisen–Sauerstoff‑Bindung über alle Zeitpunkte hinweg bei etwa 1,83 Å blieb — länger als erwartet für eine klassische Doppelbindungs‑Ferryl‑(Fe(IV)=O)‑Spezies und näher an einer Einfachbindung, während die spektralen Signaturen aus Röntgen‑Emission und optischer Absorption eindeutig hohe Oxidationszustände anzeigten, die mit Compound I und II vereinbar sind.
Einfache Erklärungen ausschließen
Da die Experimente mit ultrakurzen Pulsen bei Raumtemperatur durchgeführt wurden, konnten die üblichen Verdächtigen für verzerrte Bindungslängen — röntgeninduzierte Reduktion und kryogene Artefakte — weitgehend ausgeschlossen werden. Das Team prüfte außerdem, ob der an Eisen gebundene Sauerstoff protoniert worden sei und somit die Doppelbindung in eine hydroxidähnliche Einfachbindung verwandelt habe. Bekannte Säure‑Base‑Eigenschaften ähnlicher Hämzentren zusammen mit früheren chemischen Studien sprechen jedoch entschieden gegen eine solche Protonierung in diesem Enzymtyp. Die spektroskopischen Daten zeigten ferner, dass das Eisen nach der Reaktion mit Wasserstoffperoxid in einem hohen Oxidations‑ und niedrigspinigen Zustand verblieb, wie es für echte Ferryl‑Intermediate zu erwarten ist, was die Idee stützte, dass die unerwartet lange Bindung durch subtilere elektronische Effekte statt durch eine einfache Änderung der chemischen Form verursacht sein muss.
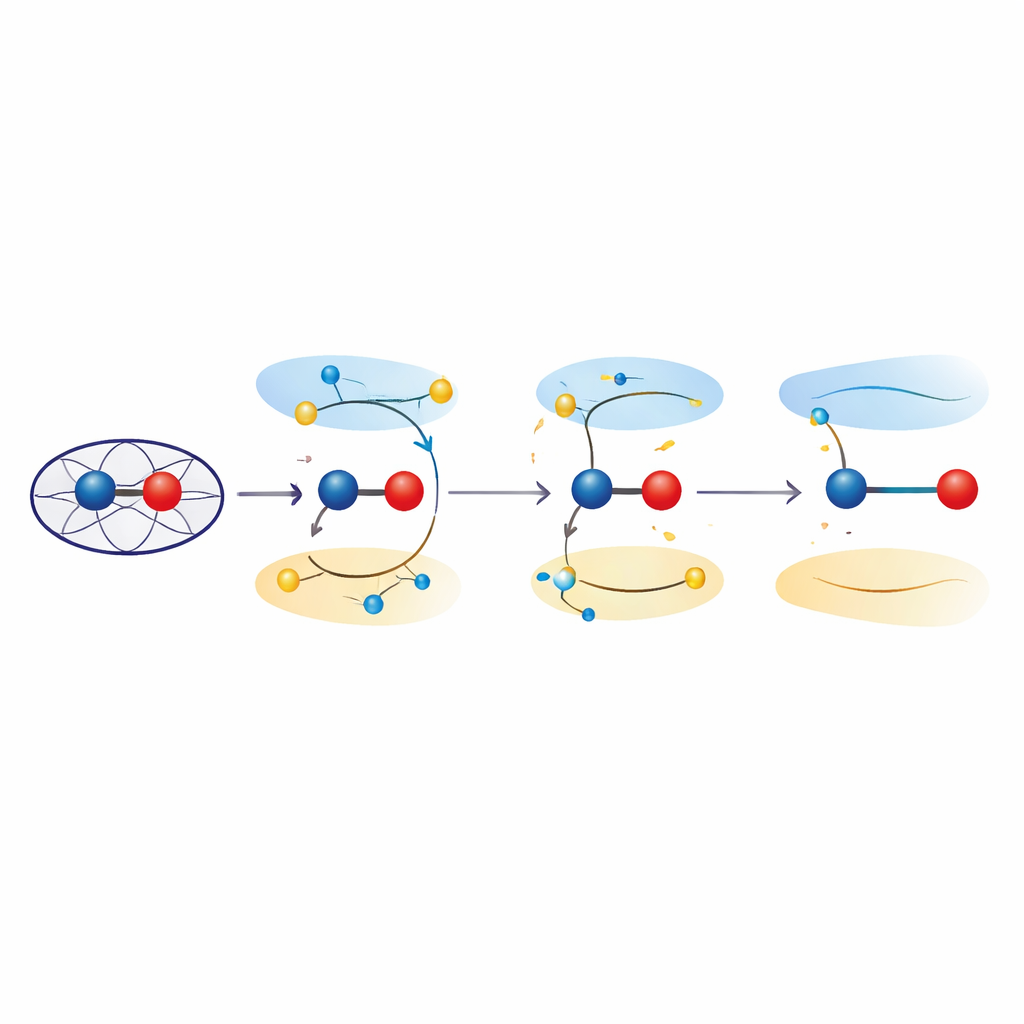
Angeregte Zustände, die Bindungen dehnen
Um diese Effekte zu untersuchen, griffen die Forscher zu quantenmechanischen Rechnungen sowohl an vereinfachten Modellen als auch in der vollständigen Proteinumgebung. Mit zeitabhängiger Dichtfunktionaltheorie und kombinierten Quantenmechanik/Molekulmechanik‑Ansätzen untersuchten sie, wie das Anheben von Elektronen aus bindenden in antibindende Orbitale in der Eisen–Sauerstoff‑Einheit die bevorzugte Bindungslänge verändert. Diese angeregten Zustände, die energetisch nahe am Grundzustand des Ferryl liegen, führten konsistent zu Eisen–Sauerstoff‑Abständen im Bereich von 1,8–1,9 Å — im Einklang mit den kristallographischen Beobachtungen. Die Analyse der Elektronenverteilung zeigte, dass dieses Paar in diesen Zuständen nicht mehr wie eine reine Fe(IV)=O‑Doppelbindung reagiert, sondern stattdessen „ferric‑oxyl“‑Charakter annimmt, mit Eigenschaften, die einem Fe(III) gebunden an ein sauerstoffzentriertes Radikal ähneln. Eine Quantenverfeinerung der experimentellen Strukturen bestätigte, dass solche angeregten Zustandsbeschreibungen die Daten mindestens ebenso gut erklären wie konventionelle Grundzustandsmodelle.
Was das für das Verständnis enzymatischer Leistung bedeutet
Kurz gesagt deutet die Arbeit darauf hin, dass lange Eisen–Sauerstoff‑Bindungen, die in diesen Häm‑Peroxidasen beobachtet werden, nicht die Annahme von Schäden, Reduktion oder versteckten Protonen erfordern. Stattdessen können sie auf natürliche Weise entstehen, wenn die Ferryl‑Einheit kurzzeitig niedrig liegende angeregte Zustände zugänglich macht, die die Bindung schwächen und ferric‑oxyl‑Charakter verleihen. Für Nicht‑Spezialisten bedeutet dies, dass das „geschäftliche Ende“ vieler sauerstoffaktivierender Enzyme dynamischer und elektronisch flexibler sein könnte, als man früher annahm — subtile Verschiebungen der Elektronenplatzierung verändern Bindungsstärke und Reaktivität, ohne die Gesamtchemie zu verändern. Die Anerkennung dieser angeregten Zustände könnte die Interpretation struktureller Daten zu starken biologischen Oxidationsmitteln neu formen und die Gestaltung künstlicher Katalysatoren leiten, die dieses empfindliche elektronische Gleichgewicht nachahmen oder gezielt einstellen.
Zitation: Williams, L.J., Kamps, J.J., Brânzanic, A.M.V. et al. Can ferric-oxyl excited states explain elongated iron-oxygen bonds in heme peroxidase catalytic intermediates?. Nat Commun 17, 2324 (2026). https://doi.org/10.1038/s41467-026-69192-8
Schlüsselwörter: Häm‑Peroxidase, Ferryl‑Intermediat, Eisen‑Sauerstoff‑Bindung, angeregte elektronische Zustände, X‑Ray Free‑Electron Laser