Clear Sky Science · de
scLong: ein Milliarden-Parameter-Basis‑Modell zur Erfassung langfristiger Genkontexte in der Einzelzell‑Transkriptomik
Computern beibringen, die verborgene Sprache der Zellen zu lesen
Jede Zelle in Ihrem Körper trägt eine geschäftige Stadt von Genen, die in komplizierten Mustern an- und abgeschaltet werden. Moderne Einzelzell‑RNA‑Sequenzierung kann inzwischen in jede einzelne Zelle „hineinhören“, doch das Ergebnis ist eine überwältigende Flut von Zahlen. Dieses Papier stellt scLong vor, ein großes KI‑Modell, das dafür entwickelt wurde, diese komplexen Genaktivitätsmuster zu verstehen, einschließlich schwacher Signale, die ältere Methoden oft übersehen. Ziel ist es, Forschern zu helfen nachzuvollziehen, wie Zellen auf das Abschalten von Genen, das Hinzufügen von Wirkstoffen oder das Auftreten von Krankheiten reagieren.
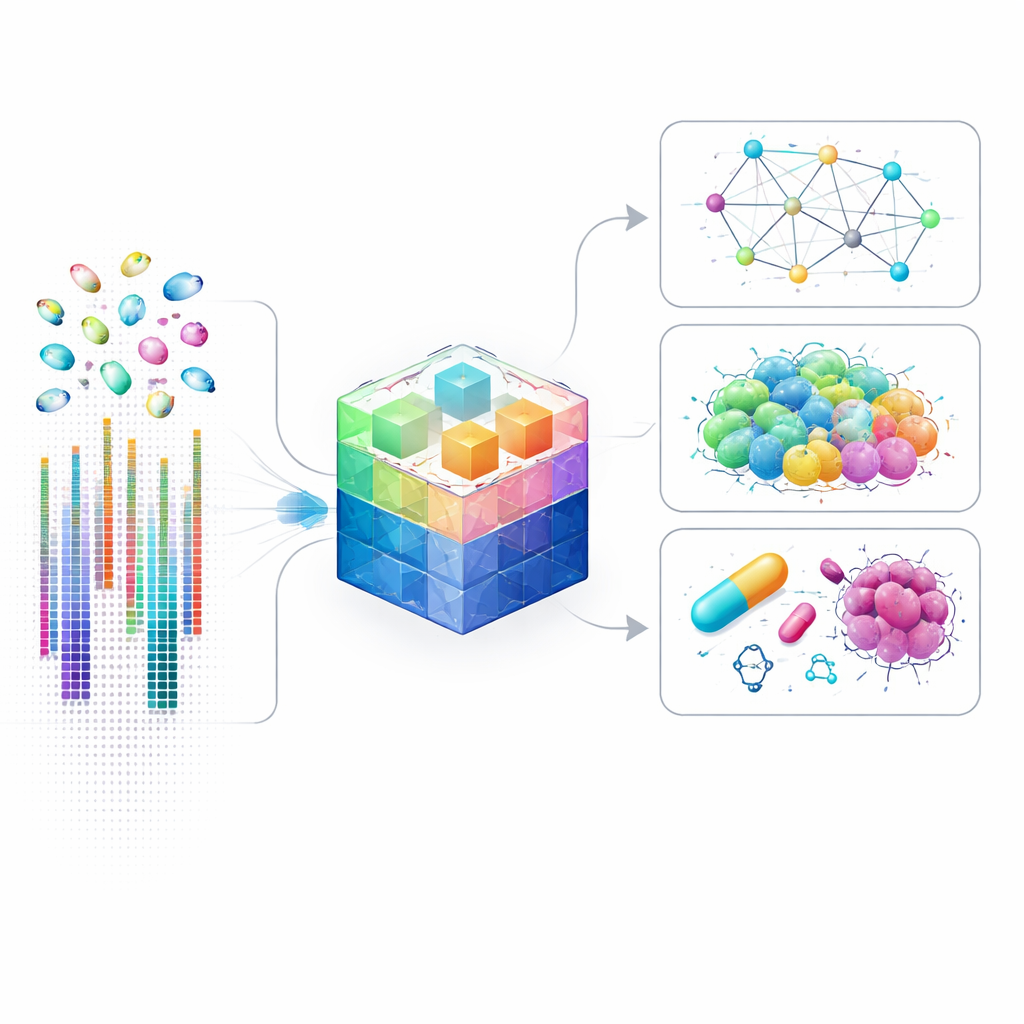
Warum Zell‑level Genkarten wichtig sind
Traditionelle Genstudien vermischen oft Millionen von Zellen, wodurch seltene oder ungewöhnliche Zelltypen ausgelöscht werden. Einzelzelltechniken haben das geändert, indem sie die Genaktivität in jeder Zelle separat messen und so versteckte Zelltypen, subtile Zell‑zu‑Zell‑Kommunikation und feingliedrige Steuerkreise sichtbar machen, die entscheiden, was eine Zelle tut. Die Analyse solcher Daten ist jedoch äußerst anspruchsvoll: Jede Zelle kann Aktivitätswerte für zehntausende Gene aufweisen, von denen viele kaum nachweisbar sind. Bestehende KI‑Modelle vereinfachen das Problem, indem sie sich nur auf die lautesten Gene konzentrieren, was die Berechnung beschleunigt, aber viele subtile Signale verpasst, die in Krankheit, Entwicklung oder Arzneimittelantwort entscheidend sein könnten.
Ein neues Modell, das auf jedes Gen hört
scLong begegnet dieser Herausforderung, indem es skaliert statt zu beschneiden. Es ist ein Milliarden‑Parameter‑Basis‑Modell, das auf Genaktivitätsprofilen von etwa 48 Millionen menschlichen Zellen aus mehr als 50 Geweben trainiert wurde. Anders als frühere Ansätze, die nur einige tausend hochaktive Gene berücksichtigen, betrachtet scLong etwa 28.000 Gene gleichzeitig, einschließlich solcher mit seltener oder schwacher Expression. Es kombiniert zwei Arten von Informationen für jedes Gen: wie aktiv es in einer gegebenen Zelle ist und was bereits über seine Funktion aus der Gene Ontology bekannt ist, einem großen, von Expert:innen kuratierten Katalog von Genrollen und Beziehungen. Ein spezialisiertes Netzwerk, das auf einem Graphen von Genverknüpfungen operiert, destilliert dieses Vorwissen in kompakte Repräsentationen, die das Modell neben den rohen Expressionswerten nutzen kann.
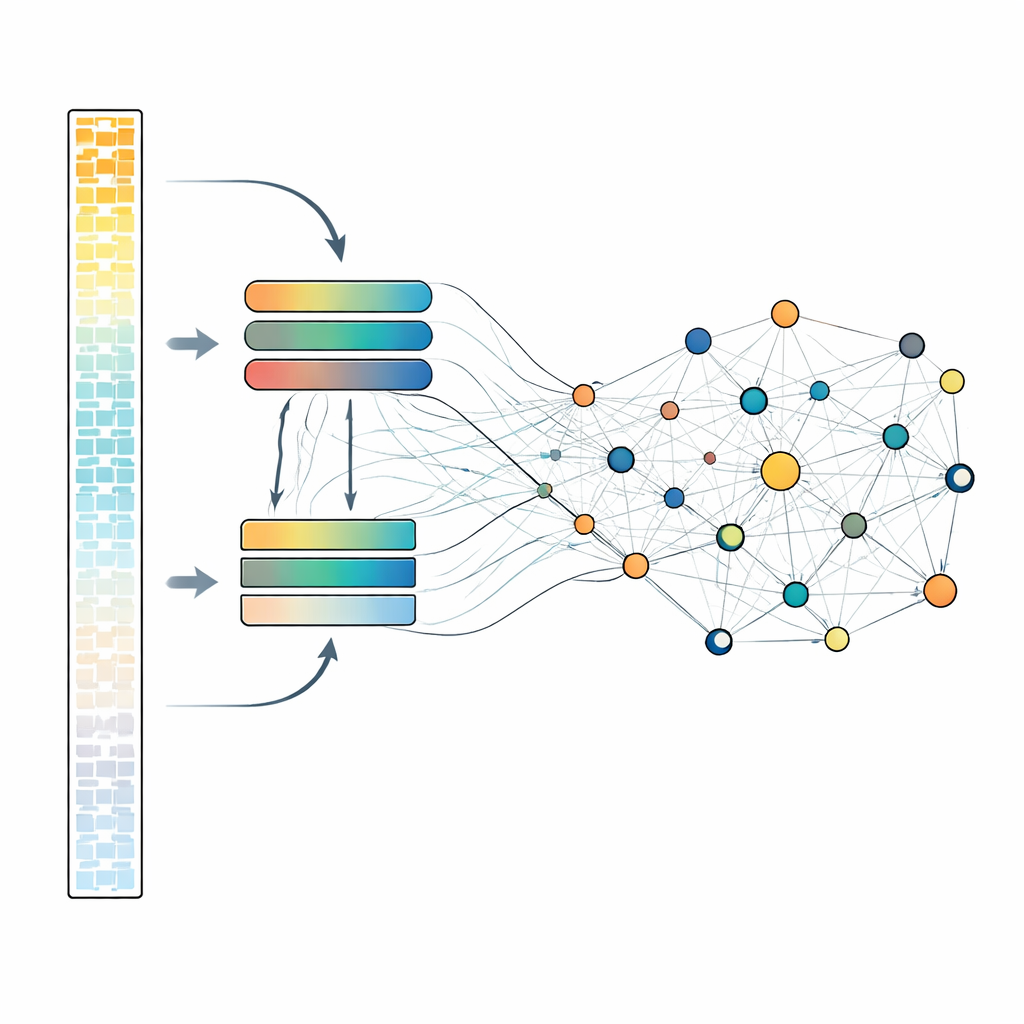
Wie das Modell Leistung und Effizienz ausbalanciert
Jedes Gen im Detail zu betrachten ist rechenaufwändig, daher verwendet scLong ein geschicktes Zwei‑Spur‑Design. Innerhalb jeder Zelle werden Gene nach ihrer Expressionsstärke sortiert. Die am stärksten aktiven Gene, die oft das wichtigste biologische Signal tragen, werden durch ein größeres, leistungsfähigeres Attention‑Modul verarbeitet. Die leiseren Gene, einschließlich niedriger oder sogar null‑Messwerte, werden durch ein kleineres, leichteres Modul geleitet. Anschließend werden alle Gene wieder zusammengeführt und durch eine weitere Attention‑Schicht geschickt, die es jedem Gen erlaubt, jedes andere zu beeinflussen. Dieses Design ermöglicht es dem Modell, kostengünstigere, aber dennoch aussagekräftige Repräsentationen für schwache Signale zu behalten, während mehr Kapazität den stärksten vorbehalten wird. Während des Pretrainings versteckt das System wiederholt einen Teil der Genaktivitätswerte und lernt, diese aus dem umgebenden Kontext zu rekonstruieren, wodurch es die Muster entdecken muss, die Gene miteinander verbinden.
Anwendung des Modells auf reale Probleme
Einmal trainiert, kann scLong an eine breite Palette biologischer Fragestellungen angepasst werden. Die Autor:innen zeigen, dass es vorhersagt, wie sich die Genaktivität verändert, wenn bestimmte Gene abgeschaltet oder verändert werden, einschließlich Kombinationen von zwei Genen, die gemeinsam wirken könnten. Es prognostiziert auch, wie Zellen auf verschiedene Chemikalien reagieren, was für Wirkstoffentdeckung und Sicherheitsprüfungen wichtig ist. In Krebsstudien hilft scLong zu antizipieren, wie Tumorzelllinien auf einzelne Wirkstoffe und auf Wirkstoffpaare reagieren, die in Kombination besser wirken könnten, und übertrifft dabei oft sowohl spezialisierte Modelle als auch andere große Basis‑Modelle. Über Vorhersagen hinaus kann scLong Netzwerke regulatorischer Beziehungen zwischen Genen ableiten und technische Verzerrungen korrigieren helfen, die entstehen, wenn Daten in verschiedenen Labors oder auf unterschiedlichen Geräten erhoben werden.
Was das für zukünftige Medizin und Forschung bedeutet
Kurz gesagt bietet scLong Wissenschaftler:innen eine hochauflösende, kontextbewusste Karte der Genaktivität in einzelnen Zellen, die leise oder selten genutzte Gene nicht verwirft. Indem es von Millionen Zellen lernt und vorhandenes biologisches Wissen einbezieht, liefert es genauere Vermutungen darüber, wie Zellen reagieren, wenn Gene gestört werden, neue Medikamente eingeführt werden oder sich Krankheitsprozesse entfalten. Das könnte die Suche nach neuen Therapien beschleunigen, personalisierte Behandlungsentscheidungen unterstützen und unser Verständnis dafür schärfen, wie komplexe Gonetzwerke Gesundheit und Krankheit steuern. Obwohl das Modell groß und rechenintensiv ist, weist es auf eine Zukunft hin, in der leistungsfähige, universell einsetzbare KI‑Systeme als vielseitige Begleiter bei der Erforschung der verborgenen Mechanismen unserer Zellen dienen.
Zitation: Bai, D., Mo, S., Zhang, R. et al. scLong: a billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics. Nat Commun 17, 2380 (2026). https://doi.org/10.1038/s41467-026-69102-y
Schlüsselwörter: Einzelzell‑Transkriptomik, Basis‑Modelle, Genregulation, Vorhersage von Arzneimittelreaktionen, Genexpression