Clear Sky Science · de
Vorhersage chemischer Ähnlichkeit zwischen Tandem-Massenspektren über Ionisationsmodi hinweg in der Metabolomik
Warum das Verbinden chemischer Punkte wichtig ist
Jeder Schluck Kaffee, jeder Atemzug und jede Medikamenteneinnahme hinterlassen winzige chemische Spuren in unserem Körper. Moderne Instrumente können Tausende dieser Moleküle gleichzeitig nachweisen, doch die Umwandlung dieser Signale in biologische Einsichten bleibt überraschend schwierig. Diese Studie stellt MS2DeepScore 2.0 vor, ein Tool auf Basis maschinellen Lernens, das Wissenschaftlern hilft, zu erkennen, wie diese Moleküle zueinander in Beziehung stehen — selbst wenn die Signale sehr unterschiedlich aufgezeichnet wurden. Dadurch verspricht es schnellere und vollständigere Interpretationen komplexer chemischer Gemische in Medizin, Ernährung und Umweltforschung.
Zwei Blickwinkel auf dasselbe Molekül
Die Massenspektrometrie ist eine zentrale Methode, die Moleküle wiegt und zerlegt, um ihre Identität offenzulegen. In Routineexperimenten messen Forschende Proben häufig in zwei Modi: einen, der positiv geladene Ionen bevorzugt, und einen für negativ geladene Ionen. Jeder Modus erzeugt seinen eigenen charakteristischen „Barcode“ von Fragmenten. Selbst wenn beide Messungen vom selben Molekül stammen, können die entstehenden Muster so unterschiedlich aussehen, dass traditionelle Vergleichsmethoden versagen. Daher analysieren Forschende die beiden Modi meist getrennt, erstellen zwei voneinander losgelöste Karten der Probe und riskieren dabei, wichtige Beziehungen zwischen Chemikalien zu übersehen.
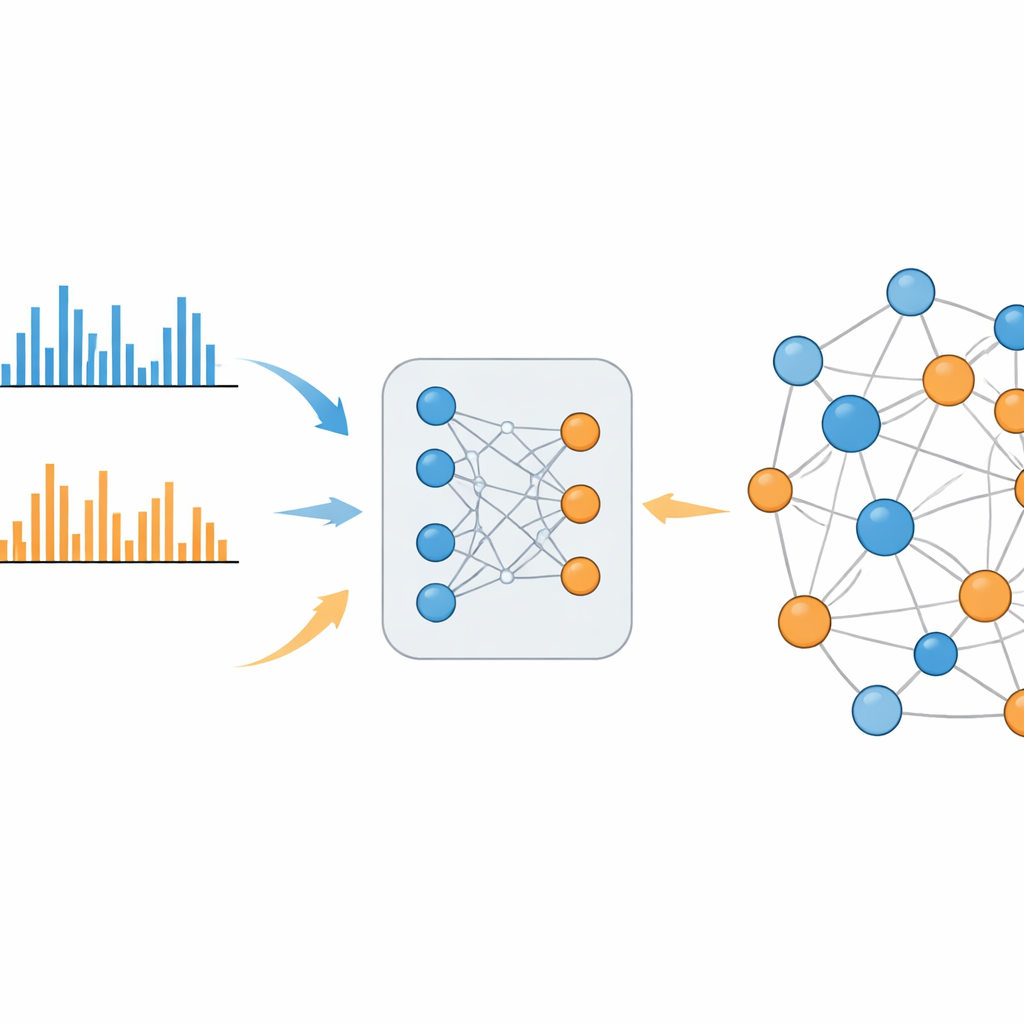
Ein lernendes System, das die Lücke schließt
MS2DeepScore 2.0 überwindet diese Trennung, indem es chemische Ähnlichkeit direkt aus großen Bibliotheken bekannter Spektren lernt. Das Modell basiert auf einer Zwillings‑Neuronalen‑Netzwerkarchitektur, die jedes Fragmentierungsmuster in einen 500‑dimensionalen Fingerabdruck, ein sogenanntes Embedding, umwandelt. Während des Trainings sieht das System Hunderttausende von Beispielen aus Positiv‑ und Negativ‑Modi sowie Angaben dazu, wie ähnlich die zugrundeliegenden Moleküle tatsächlich sind. Es passt sich so an, dass Spektren verwandter Moleküle ähnliche Embeddings erhalten — unabhängig davon, ob sie im selben oder in entgegengesetzten Modi gemessen wurden. Die neue Version erweitert das Vorgängermodell, indem zusätzliche Informationen wie die Masse des ursprünglichen Moleküls und der verwendete Ionisationsmodus eingespeist werden und indem ein sorgfältig ausbalanciertes Sampling angewandt wird, sodass seltene, aber informative chemische Beziehungen nicht von häufigen, wenig aussagekräftigen verdrängt werden.
Von verstreuten Signalen zu einheitlichen Karten
Einmal trainiert kann MS2DeepScore 2.0 abschätzen, wie chemisch ähnlich zwei Spektren sind — auch bei Paaren aus positivem und negativem Modus. Die Autoren zeigen, dass diese Vorhersagen gut mit etablierten Maßen struktureller Ähnlichkeit korrelieren, nicht nur innerhalb eines Modus, sondern auch über Modi hinweg. Anhand realer Daten aus menschlichem Urin, Blutplasma und einer wild wachsenden essbaren Pflanze bauen sie „molekulare Netzwerke“, in denen jedes Spektrum ein Knoten ist und starke vorhergesagte Ähnlichkeit eine Verbindung bildet. Im Gegensatz zu älteren Ansätzen vermischen diese Netzwerke natürlich Daten aus positiven und negativen Modi zu einer einzigen, kohärenten Karte. Von Experten kuratierte Cluster enthalten beispielsweise Gruppen koffeinverwandter Moleküle im Urin, die über Ionisationsmodi hinweg verbunden sind und bekannten Stoffwechselwegen entsprechen.
Die chemische Landschaft auf einen Blick sehen
Molekulare Netzwerke sind mächtig, können aber unübersichtlich werden, wenn zu viele schwache Verbindungen einbezogen werden. Um das zu vermeiden, verwenden die Autoren die Embeddings von MS2DeepScore direkt als Koordinaten in einer zweidimensionalen Darstellung, die mit der Technik UMAP erzeugt wurde. Jeder Punkt in dieser Karte steht für ein Spektrum, und benachbarte Punkte entsprechen Molekülen, die das Modell als chemisch ähnlich einstuft. Positiv‑ und Negativ‑Modus‑Spektren desselben Verbindungsstoffs, die visuell sehr unterschiedlich wirken, liegen in diesem Embedding‑Raum oft nebeneinander. Das Team trainiert außerdem ein zusätzliches Modell, das jedes Embedding prüft und seine Zuverlässigkeit abschätzt, sodass Spektren markiert werden, die verrauscht, unvollständig oder während des Trainings bisher nicht gesehen wurden. Das Entfernen dieser minderwertigen Punkte verbessert die Gesamtgenauigkeit und macht die Visualisierungen vertrauenswürdiger.
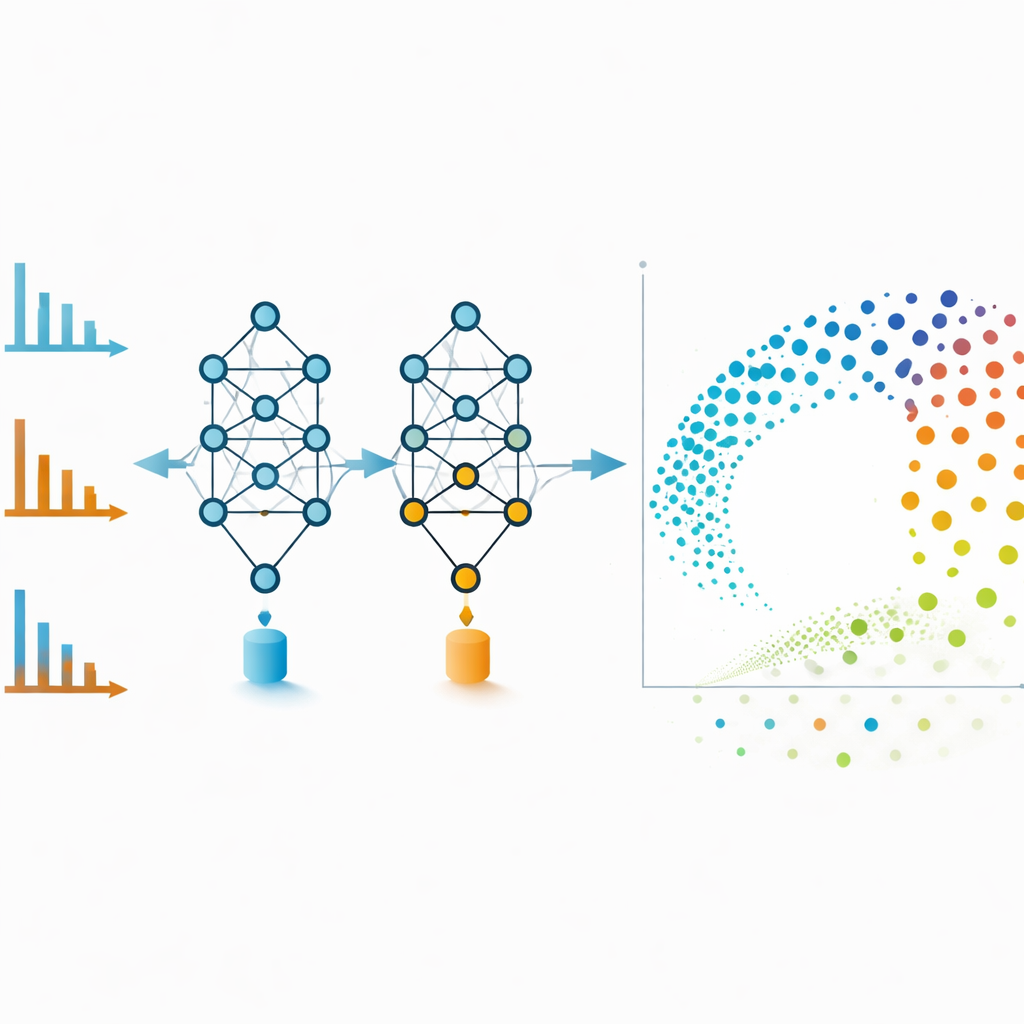
Fortgeschrittene Werkzeuge für den Alltag im Labor
Damit diese Technologie auch außerhalb von Programmier‑Experten nutzbar ist, haben die Autoren MS2DeepScore 2.0 in beliebte, frei verfügbare Massenspektrometrie‑Software integriert. Mit dieser Integration können Forschende Features erkennen, molekulare Netzwerke erstellen, die Ionisationsmodusgrenzen ignorieren, und den resultierenden chemischen Raum über interaktive Dashboards erkunden. Der Code, die trainierten Modelle und Beispieldatensätze sind offen geteilt, und das System kann für spezialisierte chemische Klassen neu trainiert oder feinabgestimmt werden.
Was das für künftige Entdeckungen bedeutet
Für Nicht‑Spezialisten lautet die Kernbotschaft: MS2DeepScore 2.0 hilft, fragmentierte und modusabhängige Messungen in ein einzelnes, verständlicheres Bild der in einer Probe vorhandenen Moleküle zu verwandeln. Indem Signale zuverlässig verknüpft werden, die früher in getrennten analytischen Welten lebten, erlaubt die Methode Forschenden, deutlich größere Referenzbibliotheken zu nutzen, Proben vollständiger zu vergleichen und sich auf sinnvolle Cluster verwandter Verbindungen zu konzentrieren. Diese Quervernetzung von Daten dürfte die Identifizierung von Biomarkern, Nährstoffen, Naturstoffen und Schadstoffen beschleunigen und letztlich unser Verständnis dafür vertiefen, wie Chemie Gesundheit und Umwelt prägt.
Zitation: de Jonge, N.F., Chekmeneva, E., Schmid, R. et al. Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics. Nat Commun 17, 2483 (2026). https://doi.org/10.1038/s41467-026-69083-y
Schlüsselwörter: Metabolomik, Massenspektrometrie, Maschinelles Lernen, molekulares Netzwerken, chemische Ähnlichkeit