Clear Sky Science · de
Kovalente Modifikation einer Glutaminsäure, inspiriert von der HaloTag‑Technologie
Proteins „Anker“ in Arzneiziele verwandeln
Viele moderne Krebsmedikamente wirken, indem sie sich an Proteine innerhalb unserer Zellen anheften. Doch einige der wichtigsten Proteine bieten keine einfachen „Griffe“, an denen Arzneistoffe halten könnten. Diese Studie stellt einen eleganten chemischen Trick vor, inspiriert von einem verbreiteten Labortool namens HaloTag, um an eine normalerweise schwer erreichbare Stelle eines Proteins zu binden, das wachstumsrelevante Signale steuert. Die Arbeit könnte neue Wege zu Medikamenten öffnen, die tumorfördernde Signalwege dauerhafter abschalten.
Warum die meisten kovalenten Medikamente auf dieselbe Stelle zielen
In den letzten Jahren haben sich sogenannte zielgerichtete kovalente Medikamente zu einer spannenden Arzneimittelklasse entwickelt. Sie tragen eine leicht reaktive chemische Gruppe, die eine permanente Bindung an eine bestimmte Aminosäure eines Proteins eingeht und das Medikament so fixiert. Fast all diese Wirkstoffe richten sich gegen Cystein, eine relativ seltene, aber sehr reaktive Aminosäure. Dagegen sind zwei andere Aminosäuren, Aspartat und Glutamat, deutlich häufiger und oft entscheidend für Form und Funktion eines Proteins, doch ihre sauren Carboxylatgruppen sind in der wässrigen Zellumgebung viel weniger reaktiv. Das macht eine selektive Modifikation schwierig, und nur wenige erfolgreiche Beispiele kovalenter Wirkstoffe, die Aspartat oder Glutamat anvisieren, gab es vor dieser Arbeit.
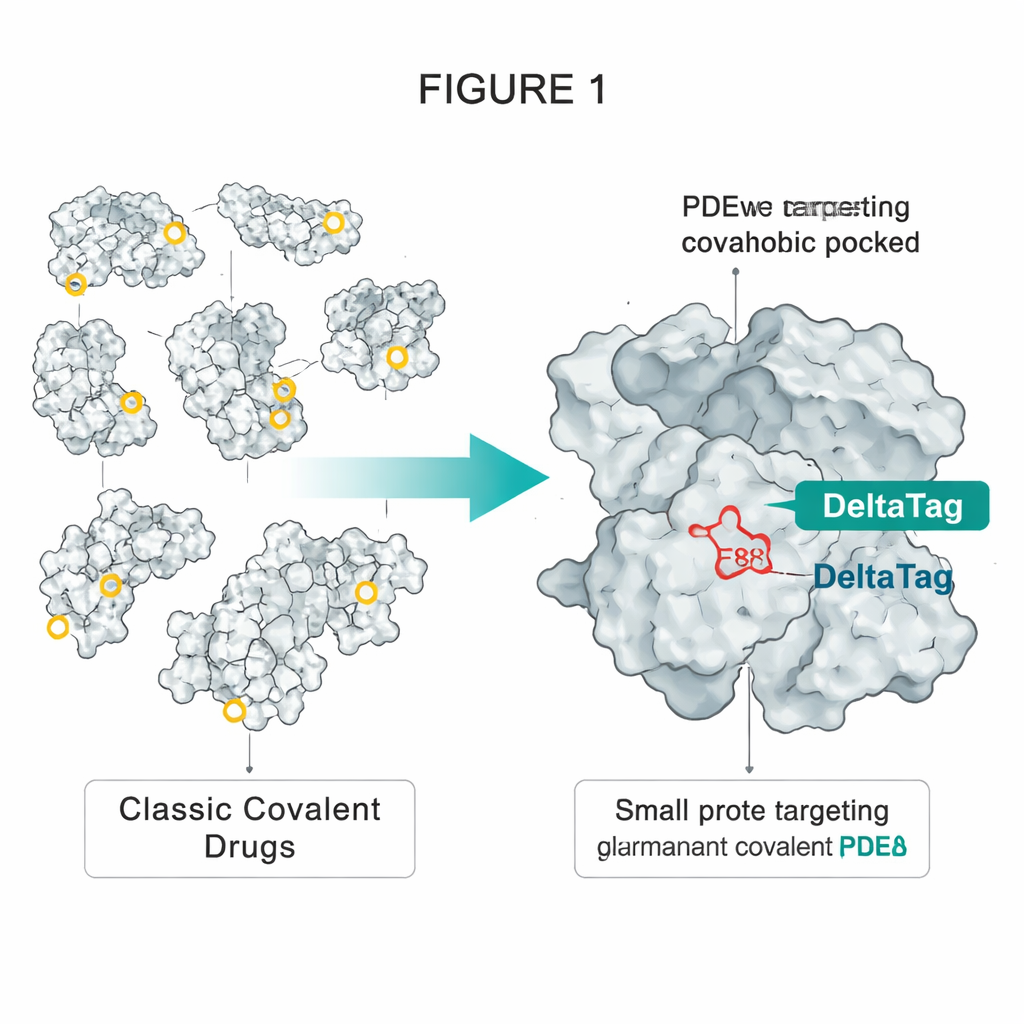
Einen Trick aus der HaloTag‑Technologie ausleihen
Die Autoren ließen sich von HaloTag inspirieren, einem weit verbreiteten, gentechnisch veränderten Protein, das dauerhaft mit fluoreszierenden Farbstoffen markiert werden kann. Bei HaloTag reagiert ein speziell positioniertes Aspartat tief in einer hydrophoben Tasche mit einer einfachen Chloroalkan‑Kette des Farbstoffs und bildet eine stabile Esterbindung. Das Team bemerkte, dass ein anderes Protein, PDEδ, eine ähnliche hydrophobe Tasche besitzt, die eine einzelne zugängliche Glutamat‑Seitenkette trägt, genannt E88. PDEδ transportiert lipidmodifizierte Signalmoleküle wie die kleine GTPase Rheb durch die Zelle und hilft ihnen, Membranen zu erreichen, wo wachstumsfördernde Komplexe wie mTORC1 aktiviert werden. Frühere nicht‑kovalente Inhibitoren von PDEδ konnten dieses Shuttling blockieren, ihre Wirkung war jedoch begrenzt, weil ein anderes Protein, Arl2, sie mit der Zeit aus der Tasche herauslösen kann.
Entwicklung von DeltaTag, um an Glutamat zu binden
Um dieses „Rauskick‑“Problem zu überwinden, begannen die Forschenden mit einem bekannten hochaffinen PDEδ‑Blocker und konstruierten an einer seiner Seitenketten ein Haloalkan‑„Warhead“, ähnlich denen, die in HaloTag‑Liganden verwendet werden. Durch mehrere Runden struktureller Anpassungen, geleitet von Protein‑Kristallstrukturen, gelangten sie zu einer Verbindung namens DeltaTag. Ihr zentrales Merkmal ist eine Phenethylbromid‑Gruppe, so positioniert, dass, sobald das Molekül in PDEδs Lipidtasche sitzt, das Bromid genau ausgerichtet ist, um mit E88 zu reagieren. Biophysikalische Messungen und hochauflösende Röntgenstrukturen bestätigten, dass DeltaTag eine kovalente Esterbindung spezifisch mit diesem Glutamat bildet und dass andere potenziell reaktivere Aminosäuren auf PDEδ unberührt bleiben. Die Verbindung ist reaktiv genug, um das Protein effizient zu markieren, aber stabil genug in Wasser und in Gegenwart des wichtigsten schwefelhaltigen Antioxidans der Zelle, Glutathion, um weitreichende, unspezifische Schäden zu vermeiden.
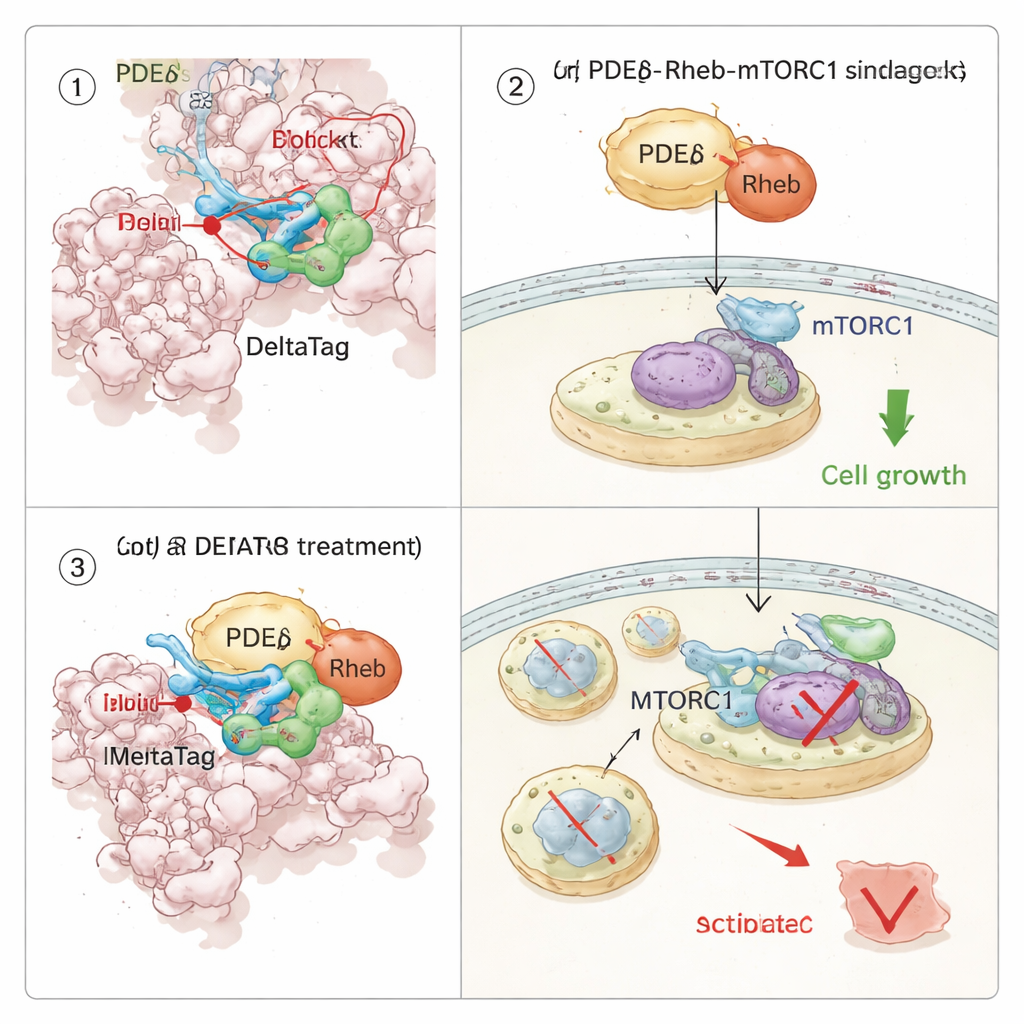
Umschaltung eines Wachstumswegs in Krebszellen
Da PDEδ Rheb chaperoniert und Rheb wiederum den mTORC1‑Wachstumspfad einschaltet, untersuchte das Team, wie sich eine kovalente Hemmung von PDEδ auf die Zellsignalgebung auswirkt. Mithilfe von thermischer Stabilitätsprofilierung über Tausende von Proteinen zeigten sie, dass DeltaTag selektiv an PDEδ in Zellextrakten bindet und Proteine im Zusammenhang mit der Rheb–mTOR‑Achse beeinflusst. In Live‑Cell‑Imaging‑Experimenten bewirkte DeltaTag, dass Rheb sich von einem diffus verteilten Muster im Zellinneren zu stärker konzentrierten internen Membranen verlagert, was mit einer Störung seines normalen Shuttlings übereinstimmt. Globale Phosphoproteomik und gezielte Western‑Blot‑Analysen zeigten, dass die mTORC1‑Aktivität, verfolgt durch die Phosphorylierung eines ribosomalen Proteins namens S6, im Zeitverlauf abnimmt, während kompensatorische Signale in verwandten Pfaden zunehmen. Wichtig ist, dass DeltaTag in mehreren menschlichen Krebszelllinien, die durch mutiertes KRAS getrieben werden und auf starke mTOR‑Signalgebung angewiesen sind, das Zellwachstum wirksamer verlangsamte als eng verwandte reversible Inhibitoren; seine Wirkung war in Zellen ohne PDEδ schwächer, was unterstützt, dass die Hauptwirkung on‑target ist.
Eine Tür zu neuen Arten von Arzneizielen öffnen
Diese Arbeit zeigt, dass es möglich ist, kleine Moleküle zu entwickeln, die eine permanente, hochspezifische Bindung an ein einzelnes Glutamat eingehen, das in einer fettigen Tasche eines Proteins verborgen ist, mithilfe einer relativ einfachen Haloalkan‑Chemie, die von HaloTag entlehnt wurde. Im Modellsystem PDEδ führt diese kovalente Verbindung zu einer dauerhafteren Blockade eines krebsrelevanten Wachstumswegs als frühere reversible Wirkstoffe. Allgemeiner könnte dieselbe Designlogik auf andere Proteine angewendet werden, die hydrophobe Hohlräume besitzen, aber die üblichen „reaktiven“ Aminosäuren vermissen, sofern sie ein strategisch gelegenes Glutamat oder Aspartat aufweisen. Mit weiterer Optimierung könnten Glutamat‑targetierende kovalente Inhibitoren wie DeltaTag nützliche Werkzeuge der chemischen Biologie und letztlich Ausgangspunkte für neue Therapien werden, die derzeit schwer angreifbare Proteine adressieren.
Zitation: Zhang, R., Liu, J., Gasper, R. et al. Covalent modification of a glutamic acid inspired by HaloTag technology. Nat Commun 17, 1257 (2026). https://doi.org/10.1038/s41467-026-68999-9
Schlüsselwörter: kovalente Inhibitoren, Glutamat‑Targeting, PDEδ, mTOR‑Signalgebung, Design von Krebsmedikamenten