Clear Sky Science · de
Entwicklungen und Herausforderungen bei der Trefferprogression in der fragmentbasierten Wirkstoffforschung
Aus winzigen chemischen Bausteinen werden zukünftige Medikamente
Moderne Medikamente werden häufig durch das Durchsieben riesiger Molekülbibliotheken entdeckt – ein Verfahren, das langsam, teuer und zunehmend ineffizient ist. Dieser Artikel beleuchtet einen neueren Ansatz, die fragmentbasierte Wirkstoffforschung, die von sehr kleinen chemischen Bausteinen ausgeht und diese Schritt für Schritt zu vielversprechenden Wirkstoffkandidaten aufbaut. Für die Leserschaft bietet er einen Einblick, wie intelligenteres Design, Automatisierung und künstliche Intelligenz die Suche nach zukünftigen Therapien beschleunigen und breiter verfügbar machen könnten.
Warum klein anfangen statt alles zu screenen
Die traditionelle Wirkstoffsuche beruht oft darauf, Millionen relativ großer Moleküle zu testen, um zu sehen, welche an ein krankheitsrelevantes Protein binden. Fragmentbasierte Methoden gehen den umgekehrten Weg: Sie screenen eine deutlich kleinere Sammlung winziger Moleküle oder „Fragmente“, die jeweils eine einfache chemische Form repräsentieren. Diese Fragmente binden nur schwach, erkunden aber aufgrund ihrer geringen Größe und Vielfalt chemische Möglichkeiten viel effizienter. Die Herausforderung besteht darin, dass schwache Signale schwer zu erkennen und zu interpretieren sind, sodass sehr empfindliche Experimente und sorgfältige Gegenkontrollen nötig sind, um sicherzustellen, dass ein Fragment tatsächlich bindet und kein Assay-Artefakt vorliegt. Strukturelle Methoden wie Röntgenkristallographie und Kryo-Elektronenmikroskopie können genau zeigen, wie ein Fragment in einer Proteintasche liegt, während Lösungsmethoden wie NMR, Kalorimetrie und Oberflächenplasmonenresonanz messen, wie stark und wie schnell es bindet.
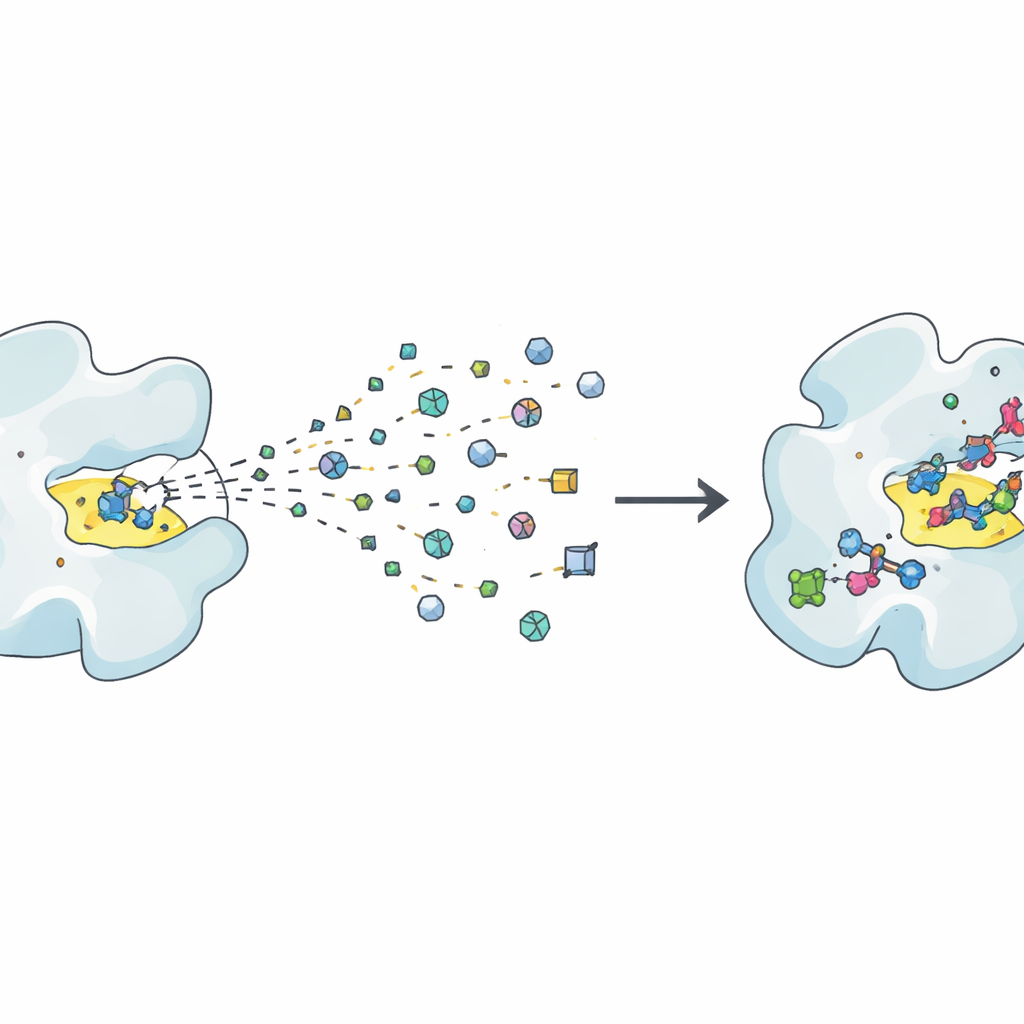
Von ersten Treffern zu vielversprechenden Leads
Wenn nützliche Fragmente gefunden sind, beginnt die eigentliche Arbeit: diese schwachen „Hits“ in starke, selektive „Lead“-Verbindungen zu verwandeln. Der Artikel beschreibt diese Reise als wiederholte „Design-Make-Test“-Zyklen. Im Design-Schritt schlagen Chemiker und Computer Wege vor, Fragmente zu vergrößern, zu verknüpfen oder zu verschmelzen, damit sie die Proteintasche besser ausfüllen, unerwünschte Reaktivität vermeiden und günstige physikalische Eigenschaften wie Löslichkeit behalten. Im Make-Schritt werden diese Entwürfe synthetisiert, zunehmend unterstützt durch Roboter, Hochdurchsatz-Chemie und clevere Routenplanungssoftware. Im Test-Schritt wird dann gemessen, ob die neuen Moleküle tatsächlich besser binden, auf die beabsichtigte biologische Funktion wirken und typische Fallstricke wie pan-assay-interfering compounds vermeiden, die Irreführung erzeugen können. Da Fragmente schwach beginnen, sind oft mehrere Runden dieses Zyklus erforderlich, bevor Verbindungen stark genug sind, um wirkliche Wirkstoffkandidaten zu ähneln.
Neue Werkzeuge: Automatisierung, KI und intelligente Bibliotheken
Die Übersicht hebt hervor, wie eine neue Generation von Werkzeugen jede Phase dieses Zyklus umgestaltet. Fragmentbibliotheken werden heute nicht nur auf Diversität ausgelegt, sondern auch darauf, „synthetisch gesellig“ zu sein – das heißt, sie lassen sich mit robusten Reaktionen leicht in viele Richtungen erweitern. Spezialisierte Fragmentsätze richten sich an bestimmte Proteinfamilien, metallhaltige Stellen oder bilden sogar kovalente Bindungen mit spezifischen Aminosäuren, was hilft, zuvor „undruggable“ Ziele anzugehen. Digital unterstützen künstliche Intelligenz-Modelle und physikbasierte Simulationen dabei, chemische Veränderungen vorzuschlagen, die die Bindung verbessern oder die Toxizität reduzieren könnten, und sie können ultra-große virtuelle Räume mit Milliarden möglicher Moleküle durchsieben. Diese Vorhersagen werden zunehmend mit Active-Learning-Schleifen kombiniert, in denen eine kleine Anzahl teurer Simulationen oder Experimente schnellere Modelle trainiert, die die nächste Welle von Entwürfen steuern können.
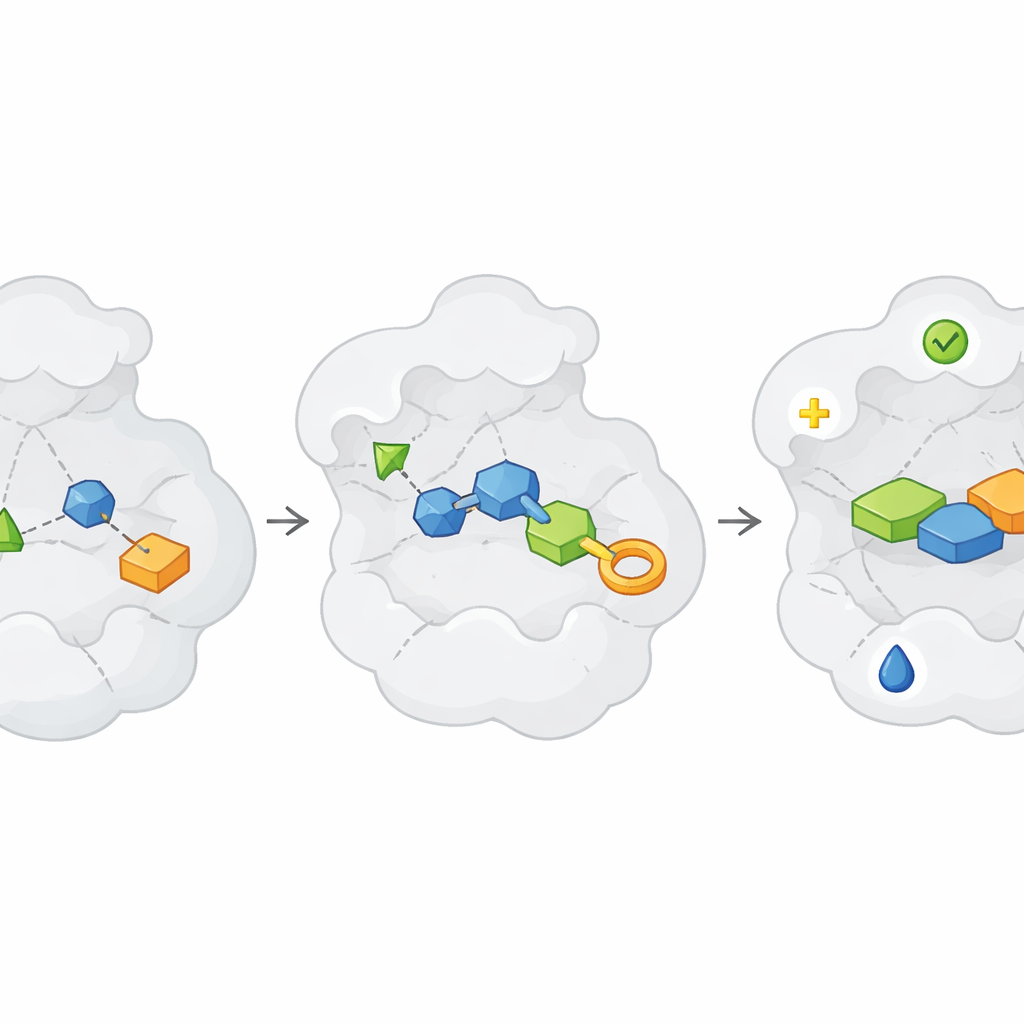
Herstellen und Testen im großen Maßstab ohne Verzögerung
Ein maßgeblicher Engpass in der Wirkstoffforschung ist schlicht das Herstellen und Reinigen ausreichend vieler Verbindungen zum Testen. Der Artikel beschreibt, wie Hochdurchsatz-Syntheseroboter, Durchflusschemie und neue Extraktionsmethoden Hunderte oder Tausende verwandter Moleküle rund um einen Fragment-Treffer erzeugen können. Einige Ansätze verzichten zunächst sogar auf eine vollständige Reinigung: rohe Reaktionsmischungen werden direkt in empfindlichen Assays wie Kristallographie, kinetischen Messungen oder NMR getestet – ein Ansatz, der manchmal als „direct-to-biology“ bezeichnet wird. Qualitätskontrollen wie Massenspektrometrie werden parallel eingesetzt, um nachzuvollziehen, welche Mischungen tatsächlich das beabsichtigte Produkt enthalten. Obwohl die Daten lärmbehaftet sein können, ermöglicht die Kombination dieser schnellen Tests mit smarten Analysen und anschließender sauberer Synthese den Forschern, Struktur–Aktivitäts-Beziehungen viel schneller abzubilden als mit traditioneller Einzel-mit-Einzel-Chemie.
Was das für künftige Medikamente bedeutet
Insgesamt kommt der Artikel zu dem Schluss, dass die fragmentbasierte Wirkstoffforschung zu einer mächtigen und flexiblen Strategie für die Entdeckung neuer Medikamente gereift ist, besonders in Verbindung mit moderner Automatisierung und KI. Vom Start mit winzigen, effizienten Bausteinen aus lässt sich der chemische Raum durchdachter erkunden, erfordert aber sorgfältige Validierung, weil die Anfangssignale so schwach sind. Die Autoren argumentieren, dass die größten Gewinne aus der engen Integration von Design, Synthese und Test in halbautomatisierte, datengetriebene Workflows sowie aus offenem Teilen von Fragmentdaten und -methoden entstehen werden, damit auch ressourcenärmere Gruppen profitieren können. Wenn diese Entwicklungen anhalten, könnten fragmentbasierte Ansätze dazu beitragen, den langfristigen Rückgang der Forschungsproduktivität umzukehren und die Ankunft sichererer, wirksamerer Medikamente für eine breite Palette von Krankheiten zu beschleunigen.
Zitation: Grosjean, H., Biggin, P.C. Developments and challenges in hit progression within fragment-based drug discovery. Nat Commun 17, 2226 (2026). https://doi.org/10.1038/s41467-026-68941-z
Schlüsselwörter: fragmentbasierte Wirkstoffforschung, Hit-to-Lead-Optimierung, Design-Make-Test-Zyklus, Hochdurchsatz-Screening, computergestütztes Wirkstoffdesign