Clear Sky Science · de
Menschliche iPSC-basierte Modellierung der Lungenfibrose zeigt, dass p300/CBP‑Hemmung den alveolären Übergangszustand unterdrückt
Warum vernarbte Lungen uns alle betreffen
Die idiopathische Lungenfibrose (IPF) ist eine unaufhaltsame Erkrankung, bei der Lungengewebe allmählich zu Narbengewebe umgebaut wird, sodass jeder Atemzug schwerer fällt. Die derzeit verfügbaren Medikamente können diesen Prozess nur verlangsamen und verursachen oft belastende Nebenwirkungen. In dieser Studie nutzten die Forschenden moderne Stammzell‑ und Genomik‑Werkzeuge, um vernarbte Lungen im Labor nachzubilden und eine einfache, aber wichtige Frage zu stellen: Gibt es einen Schalter, der beschädigte Lungenzellen aus einem schädlichen Zustand herausführt und sie zurück in Richtung Heilung lenkt?
Ein im Labor gezüchtetes Fenster in die vernarbte Lunge
Um IPF zu untersuchen, bauten die Forschenden miniaturisierte Lungen aus menschlichen induzierten pluripotenten Stammzellen (iPSZ). Diese iPSZ wurden so gelenkt, dass sie zu alveolären Zellen wurden — den Zellen, die die winzigen Lungenbläschen auskleiden, in denen Sauerstoff ins Blut gelangt — und zusammen mit Lungenfibroblasten, den Bindegewebszellen, die Narben bilden, kultiviert. Eingebettet in ein weiches Gel verhielten sich diese „alveolären Organoide“ ähnlich wie echtes Lungengewebe. Bei Exposition gegenüber dem Chemotherapeutikum Bleomycin, einem bekannten Auslöser von Lungenschäden, schrumpften die Gele, weil Fibroblasten an ihnen zogen, was die Gewebekontraktion nachahmte, die bei Fibrose beobachtet wird.
Mit diesem System screente das Team eine Bibliothek von 264 kleinen Molekülen und maß automatisch, wie stark jedes Medikament die Gelkontraktion verhinderte, wobei ein Deep‑Learning‑Bildanalyse‑Tool für objektive Auslesungen sorgte. Viele Verbindungen zeigten keinen Effekt, aber eine Klasse stach deutlich hervor: Hemmer der Proteine p300 und CBP, die mitbestimmen, wie DNA verpackt ist und welche Gene ein‑ oder ausgeschaltet werden. Alle acht p300/CBP‑gerichteten Verbindungen in der Bibliothek reduzierten die Kontraktion in niedrigen Dosen und hoben diesen Signalweg als vielversprechenden Ansatzpunkt gegen Fibrose hervor. 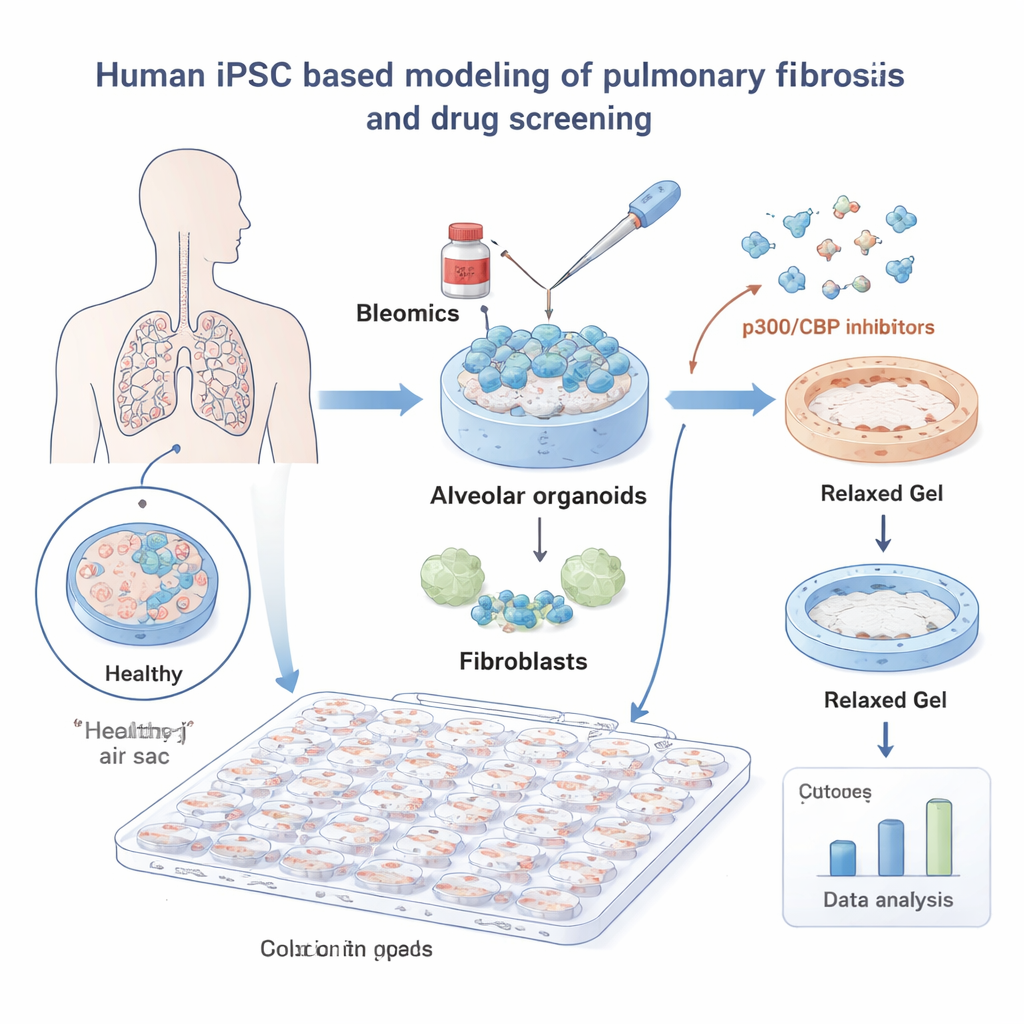
Die Übeltäter: transitorische Lungenzellen
Jüngere Arbeiten haben einen problematischen „Zwischen“-Zelltyp in erkrankten Lungen aufgedeckt, den alveolären transitorischen Zellzustand. Normalerweise reifen Stützzellen, sogenannte AT2‑Zellen, zu ultradünnen AT1‑Zellen heran, die die Lungenbläschen bedecken und den Gasaustausch ermöglichen. Bei IPF bleiben AT2‑Zellen jedoch häufig in diesem transitorischen Zustand stecken, exprimieren Stress‑ und Reparaturgene und schaffen es nicht, die Umwandlung zu voll funktionsfähigen AT1‑Zellen abzuschließen. Diese transitorischen Zellen sammeln sich in fibrotischen Bereichen und kommunizieren stark mit Fibroblasten, doch bisher war unklar, ob sie bloße Schadensfolgen oder aktive Treiber der Vernarbung sind.
Durch RNA‑Sequenzierung und Profilierung offener Chromatinregionen in ihren Organoiden zeigten die Autorinnen und Autoren, dass die in ihrem Modell entstehenden transitorischen Zellen jenen in Lungen von IPF‑Patienten eng entsprachen. Diese induzierten transitorischen Zellen zeigten Gen‑Signaturen von Stress, Entzündung und Matrix‑Remodellierung und aktivierten die ko‑kultivierten erwachsenen Lungenfibroblasten stark. Entscheidenderweise sanken bei Blockade von p300/CBP die Marker des transitorischen Zustands, die AT2‑Identität blieb besser erhalten und die Aktivierung der Fibroblasten ließ nach. Anders gesagt: Die Wirkstoffe vergifteten Zellen nicht allgemein; sie verhinderten gezielt, dass AT2‑Zellen in dieses schädliche Zwischenstadium geraten.
Die molekularen Schaltkreise entwirren
Um zu verstehen, wie p300/CBP diese Schicksalsentscheidung beeinflusst, untersuchte das Team chemische Markierungen auf Histonen — Proteinen, die helfen, DNA zu verpacken. Eine bestimmte Markierung, die Acetylierung von H3K27, wird häufig von p300/CBP an aktiven Enhancern und Promotoren angebracht. In transitorischen Zellen wiesen Regionen in der Nähe von Stress‑Antwort‑ und pro‑fibrotischen Genen starke H3K27‑Acetylierungen auf und waren angereichert für Bindungsstellen von Transkriptionsfaktoren wie AP‑1 und HNF1B. Nach Behandlung mit p300/CBP‑Inhibitoren gingen diese Acetyl‑Markierungen an diesen Stellen zurück, und die Expression vieler pro‑fibrotischer Gene fiel. Auch die direkte Blockade von AP‑1 oder die Reduktion von AP‑1 und HNF1B mittels kleiner interferierender RNAs dämpfte das transitorische Programm und die Organoid‑Kontraktion, was dieses Trio — p300/CBP, AP‑1 und HNF1B — als Motor der fibrotischen Umgestaltung verknüpft.
Über das Zellkultursystem hinaus testete die Studie einen Inhibitor, CBP30, in Mäusen mit Bleomycin‑induzierter Lungenverletzung. Tieren, die CBP30 erhielten, zeigten weniger transitorische Epithelzellen, geringere Aktivierung narbenbildender Myofibroblasten und reduzierte Expression von Fibrose‑Markern. Diese Kreuzvalidierung zwischen humanen Stammzellmodellen und einem Tiermodell stärkt die Annahme, dass p300/CBP kein bloßes Laborartefakt, sondern ein echter Regulator der Lungenvernarbung ist. 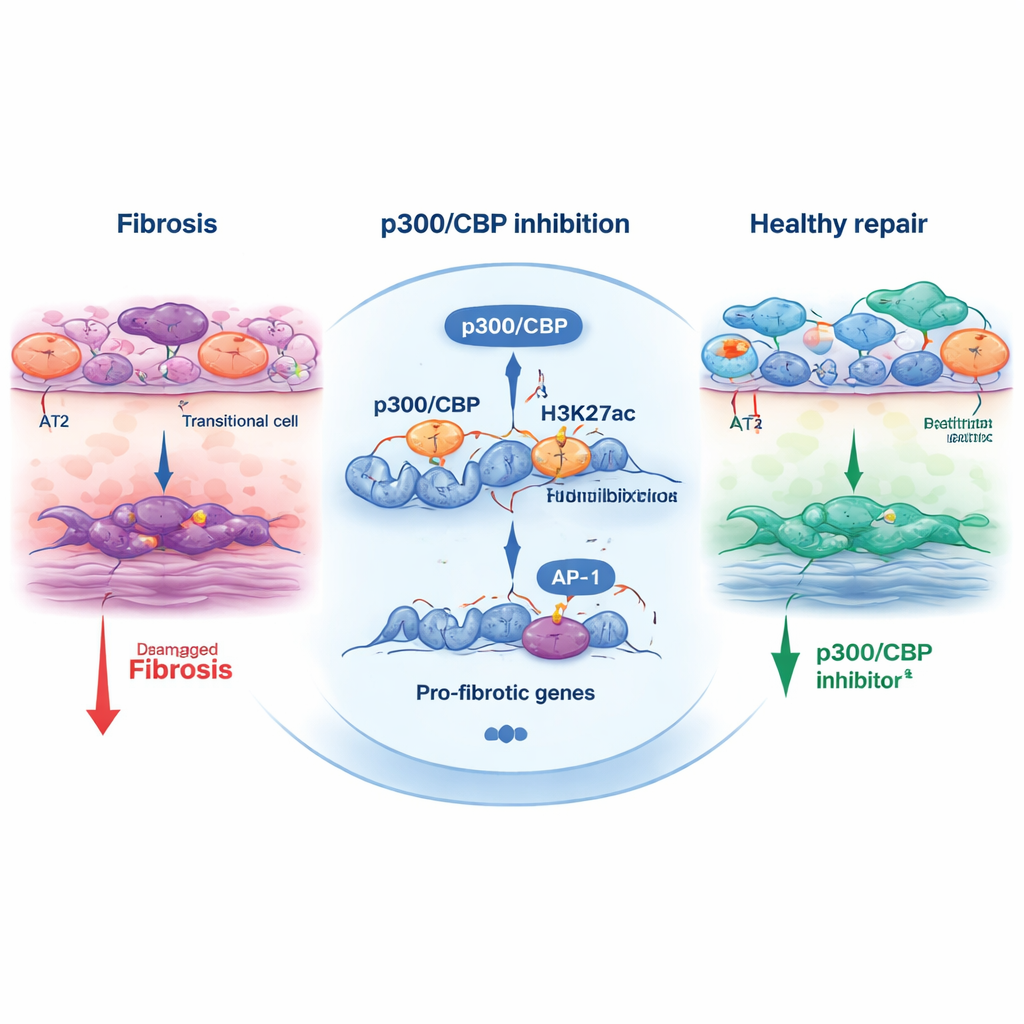
Was das für künftige Behandlungen bedeutet
Für alle Nicht‑Fachleute lautet die Kernbotschaft: Die Autorinnen und Autoren haben ein realistisches humanes Modell fibrotischer Lungen entwickelt und damit ein neues Therapieziel ins Blickfeld gerückt. Ihre Arbeit legt nahe, dass Lungenvernarbung zum Teil von einem reversiblen, stressinduzierten transitorischen Zellzustand getrieben wird, der das umliegende Gewebe fehlleitet. Durch das Herunterregeln von p300/CBP könnte man diesen Zustand beruhigen, alveoläre Zellen auf einem gesunden Entwicklungsweg halten und die Signale reduzieren, die Fibroblasten in den Überantrieb treiben. Obwohl p300/CBP‑Inhibitoren noch in puncto Sicherheit optimiert und klinisch erprobt werden müssen, weist diese Studie auf Therapien hin, die die zugrunde liegende zelluläre Fehlkommunikation bei IPF angehen, statt nur deren Folgen zu verlangsamen.
Zitation: Tsutsui, Y., Masui, A., Konishi, S. et al. Human iPSC-based Modeling of Pulmonary Fibrosis Reveals p300/CBP Inhibition Suppresses Alveolar Transitional Cell State. Nat Commun 17, 1214 (2026). https://doi.org/10.1038/s41467-026-68909-z
Schlüsselwörter: idiopathische Lungenfibrose, alveoläre Organoide, p300/CBP‑Inhibitoren, transitorische Epithelzellen, Lungenstammzellen